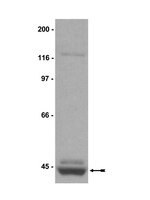
Reduced methylation of PFKFB3 in cancer cells shunts glucose towards the pentose phosphate pathway.
Yamamoto, T; Takano, N; Ishiwata, K; Ohmura, M; Nagahata, Y; Matsuura, T; Kamata, A; Sakamoto, K; Nakanishi, T; Kubo, A; Hishiki, T; Suematsu, M
Nature communications
5
3480
2014
Show Abstract
Haem oxygenase (HO)-1/carbon monoxide (CO) protects cancer cells from oxidative stress, but the gas-responsive signalling mechanisms remain unknown. Here we show using metabolomics that CO-sensitive methylation of PFKFB3, an enzyme producing fructose 2,6-bisphosphate (F-2,6-BP), serves as a switch to activate phosphofructokinase-1, a rate-limiting glycolytic enzyme. In human leukaemia U937 cells, PFKFB3 is asymmetrically di-methylated at R131 and R134 through modification by protein arginine methyltransferase 1. HO-1 induction or CO results in reduced methylation of PFKFB3 in varied cancer cells to suppress F-2,6-BP, shifting glucose utilization from glycolysis toward the pentose phosphate pathway. Loss of PFKFB3 methylation depends on the inhibitory effects of CO on haem-containing cystathionine β-synthase (CBS). CBS modulates remethylation metabolism, and increases NADPH to supply reduced glutathione, protecting cells from oxidative stress and anti-cancer reagents. Once the methylation of PFKFB3 is reduced, the protein undergoes polyubiquitination and is degraded in the proteasome. These results suggest that the CO/CBS-dependent regulation of PFKFB3 methylation determines directional glucose utilization to ensure resistance against oxidative stress for cancer cell survival. | | | 24633012
 |
Automethylation of protein arginine methyltransferase 8 (PRMT8) regulates activity by impeding S-adenosylmethionine sensitivity.
Dillon, MB; Rust, HL; Thompson, PR; Mowen, KA
The Journal of biological chemistry
288
27872-80
2013
Show Abstract
Protein arginine methyltransferase (PRMT) 8 is unique among the PRMTs, as it has a highly restricted tissue expression pattern and an N terminus that contains two automethylation sites and a myristoylation site. PRMTs catalyze the transfer of a methyl group from S-adenosylmethionine (AdoMet) to a peptidylarginine on a protein substrate. Currently, the physiological roles, regulation, and cellular substrates of PRMT8 are poorly understood. However, a thorough understanding of PRMT8 kinetics should provide insights into each of these areas, thereby enhancing our understanding of this unique enzyme. In this study, we determined how automethylation regulates the enzymatic activity of PRMT8. We found that preventing automethylation with lysine mutations (preserving the positive charge of the residue) increased the turnover rate and decreased the Km of AdoMet but did not affect the Km of the protein substrate. In contrast, mimicking automethylation with phenylalanine (i.e. mimicking the increased hydrophobicity) decreased the turnover rate. The inhibitory effect of the PRMT8 N terminus could be transferred to PRMT1 by creating a chimeric protein containing the N terminus of PRMT8 fused to PRMT1. Thus, automethylation of the N terminus likely regulates PRMT8 activity by decreasing the affinity of the enzyme for AdoMet. | Western Blotting | | 23946480
|
A versatile method to design stem-loop primer-based quantitative PCR assays for detecting small regulatory RNA molecules.
Czimmerer, Z; Hulvely, J; Simandi, Z; Varallyay, E; Havelda, Z; Szabo, E; Varga, A; Dezso, B; Balogh, M; Horvath, A; Domokos, B; Torok, Z; Nagy, L; Balint, BL
PloS one
8
e55168
2013
Show Abstract
Short regulatory RNA-s have been identified as key regulators of gene expression in eukaryotes. They have been involved in the regulation of both physiological and pathological processes such as embryonal development, immunoregulation and cancer. One of their relevant characteristics is their high stability, which makes them excellent candidates for use as biomarkers. Their number is constantly increasing as next generation sequencing methods reveal more and more details of their synthesis. These novel findings aim for new detection methods for the individual short regulatory RNA-s in order to be able to confirm the primary data and characterize newly identified subtypes in different biological conditions. We have developed a flexible method to design RT-qPCR assays that are very sensitive and robust. The newly designed assays were tested extensively in samples from plant, mouse and even human formalin fixed paraffin embedded tissues. Moreover, we have shown that these assays are able to quantify endogenously generated shRNA molecules. The assay design method is freely available for anyone who wishes to use a robust and flexible system for the quantitative analysis of matured regulatory RNA-s. | Western Blotting | | 23383094
|
Protective actions of nebivolol on chronic nitric oxide synthase inhibition-induced hypertension and chronic kidney disease in the rat: a comparison with angiotensin II receptor blockade.
Moningka, NC; Tsarova, T; Sasser, JM; Baylis, C
Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association
27
913-20
2012
Show Abstract
Nitric oxide (NO) deficiency contributes to chronic kidney disease (CKD) progression and hypertension. The β-blocker, nebivolol (N), also enhances NO production, and we studied whether N attenuates CKD and hypertension caused by chronic NO synthase inhibition (CNOSI).Male Sprague-Dawley rats on 6 weeks of CNOSI (L-NAME, 150 mg/L drinking water) received placebo (P), N (10 mg/kg/day), olmesartan (O, 2.5 mg/kg/day) or N + O. Blood pressure (BP) and urine protein and NOx (metabolites of NO) were monitored throughout. We measured glomerular sclerosis (GS), creatinine clearance (C(Cr)) and components of the NO and oxidant pathways in the renal cortex.BP increased greater than 50 mmHg in P by weeks 4-6, but no change occurred in N, O or N + O. P rats developed proteinuria and GS and C(Cr) was ∼30% of normal. In N, O and N + O, all values remained normal. In renal cortex of P, p22phox and nitrotyrosine abundance as well as H(2)O(2) levels were higher and extracellular superoxide dismutase (EC SOD) was lower versus normal kidneys. N, O and N + O normalized p22phox, H(2)O(2) and EC SOD and increased Mn SOD above normal. The cortical neuronal NO synthase (nNOS) β abundance increased in P and this was prevented by N, O and N + O.We suggest that the major benefit from both N and O is reduction in oxidative stress in the renal cortex, which may potentiate residual local NO. There was no additive benefit of N + O since each drug effectively prevented injury, but a combination may be beneficial where protection is incomplete with each drug. The increased nNOSβ protein seen early in the course of the CKD may contribute to the evolving GS. | | | 21856762
|
PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential.
Shia, WJ; Okumura, AJ; Yan, M; Sarkeshik, A; Lo, MC; Matsuura, S; Komeno, Y; Zhao, X; Nimer, SD; Yates, JR; Zhang, DE
Blood
119
4953-62
2012
Show Abstract
Fusion protein AML1-ETO, resulting from t(8;21) translocation, is highly related to leukemia development. It has been reported that full-length AML1-ETO blocks AML1 function and requires additional mutagenic events to promote leukemia. We have previously shown that the expression of AE9a, a splice isoform of AML1-ETO, can rapidly cause leukemia in mice. To understand how AML1-ETO is involved in leukemia development, we took advantage of our AE9a leukemia model and sought to identify its interacting proteins from primary leukemic cells. Here, we report the discovery of a novel AE9a binding partner PRMT1 (protein arginine methyltransferase 1). PRMT1 not only interacts with but also weakly methylates arginine 142 of AE9a. Knockdown of PRMT1 affects expression of a specific group of AE9a-activated genes. We also show that AE9a recruits PRMT1 to promoters of AE9a-activated genes, resulting in enrichment of H4 arginine 3 methylation, H3 Lys9/14 acetylation, and transcription activation. More importantly, knockdown of PRMT1 suppresses the self-renewal capability of AE9a, suggesting a potential role of PRMT1 in regulating leukemia development. | | | 22498736
|
Nebivolol does not protect against 5/6 ablation/infarction induced chronic kidney disease in rats - comparison with angiotensin II receptor blockade.
Sasser, JM; Moningka, NC; Tsarova, T; Baylis, C
Life sciences
91
54-63
2012
Show Abstract
Nitric oxide (NO) deficiency contributes to chronic kidney disease progression. Nebivolol, a beta adrenergic receptor antagonist, may enhance endogenous NO. Here, we investigated whether Nebivolol attenuates hypertension and renal injury after 5/6 ablation/infarction (A/I). Efficacy was compared to the AT1 receptor antagonist Olmesartan.Kidney disease and hypertension were induced by right kidney ablation and ~2/3 infarction of the left kidney. Rats were treated orally with vehicle (placebo), Nebivolol (5mg/kg b.i.d.), or Olmesartan (2.5mg/kg/day) for 6 weeks after A/I.With placebo, glomerular sclerosis and tubulointersititial fibrosis developed with increased blood pressure and proteinuria, and a fall in NO(x) excretion. Olmesartan prevented these changes, but Nebivolol had no effect on these measures but lowered heart rate. Neither treatment reduced systemic oxidative stress (urinary hydrogen peroxide and TBARS). Compared to controls, renal cortex abundance of nNOSα decreased and nNOSβ increased in rats after 5/6 A/I, with no changes in eNOS. Neither treatment restored nNOSα; however, both reduced nNOSβ. Activity of DDAH was decreased by 5/6 A/I but restored by both treatments despite no increase in DDAH protein abundance. Kidney cortex abundance of manganese SOD fell after 5/6 A/I and was restored by treatment with Olmesartan but not Nebivolol. Extracellular and copper/zinc SOD abundances were not changed.In conclusion, Nebivolol showed no benefit after 6 weeks in rapidly progressing, ANG II-dependent 5/6 A/I model of chronic kidney disease. This contrasts to the protection seen with 6 month treatment of Nebivolol in the slowly progressing 5/6 ablation model. | Western Blotting | | 22727796
|
Protein arginine methyltransferase 7 regulates cellular response to DNA damage by methylating promoter histones H2A and H4 of the polymerase δ catalytic subunit gene, POLD1.
Karkhanis, V; Wang, L; Tae, S; Hu, YJ; Imbalzano, AN; Sif, S
The Journal of biological chemistry
287
29801-14
2012
Show Abstract
Covalent modification of histones by protein arginine methyltransferases (PRMTs) impacts genome organization and gene expression. In this report, we show that PRMT7 interacts with the BRG1-based hSWI/SNF chromatin remodeling complex and specifically methylates histone H2A Arg-3 (H2AR3) and histone H4 Arg-3 (H4R3). To elucidate the biological function of PRMT7, we knocked down its expression in NIH 3T3 cells and analyzed global gene expression. Our findings show that PRMT7 negatively regulates expression of genes involved in DNA repair, including ALKBH5, APEX2, POLD1, and POLD2. Chromatin immunoprecipitation (ChIP) revealed that PRMT7 and dimethylated H2AR3 and H4R3 are enriched at target DNA repair genes in parental cells, whereas PRMT7 knockdown caused a significant decrease in PRMT7 recruitment and H2AR3/H4R3 methylation. Decreased PRMT7 expression also resulted in derepression of target DNA repair genes and enhanced cell resistance to DNA-damaging agents. Furthermore, we show that BRG1 co-localizes with PRMT7 on target promoters and that expression of a catalytically inactive form of BRG1 results in derepression of PRMT7 target DNA repair genes. Remarkably, reducing expression of individual PRMT7 target DNA repair genes showed that only the catalytic subunit of DNA polymerase, POLD1, was able to resensitize PRMT7 knock-down cells to DNA-damaging agents. These results provide evidence for the important role played by PRMT7 in epigenetic regulation of DNA repair genes and cellular response to DNA damage. | | | 22761421
|
Five friends of methylated chromatin target of protein-arginine-methyltransferase[prmt]-1 (chtop), a complex linking arginine methylation to desumoylation.
Fanis, P; Gillemans, N; Aghajanirefah, A; Pourfarzad, F; Demmers, J; Esteghamat, F; Vadlamudi, RK; Grosveld, F; Philipsen, S; van Dijk, TB
Molecular & cellular proteomics : MCP
11
1263-73
2012
Show Abstract
Chromatin target of Prmt1 (Chtop) is a vertebrate-specific chromatin-bound protein that plays an important role in transcriptional regulation. As its mechanism of action remains unclear, we identified Chtop-interacting proteins using a biotinylation-proteomics approach. Here we describe the identification and initial characterization of Five Friends of Methylated Chtop (5FMC). 5FMC is a nuclear complex that can only be recruited by Chtop when the latter is arginine-methylated by Prmt1. It consists of the co-activator Pelp1, the Sumo-specific protease Senp3, Wdr18, Tex10, and Las1L. Pelp1 functions as the core of 5FMC, as the other components become unstable in the absence of Pelp1. We show that recruitment of 5FMC to Zbp-89, a zinc-finger transcription factor, affects its sumoylation status and transactivation potential. Collectively, our data provide a mechanistic link between arginine methylation and (de)sumoylation in the control of transcriptional activity. | Western Blotting | Human | 22872859
|
Arginine methylation of BCL-2 antagonist of cell death (BAD) counteracts its phosphorylation and inactivation by Akt.
Sakamaki, J; Daitoku, H; Ueno, K; Hagiwara, A; Yamagata, K; Fukamizu, A
Proceedings of the National Academy of Sciences of the United States of America
108
6085-90
2011
Show Abstract
Protein arginine methylation is a common posttranslational modification catalyzed by a family of the protein arginine methyltransferases (PRMTs). We have previously reported that PRMT1 methylates Forkhead box O transcription factors at two arginine residues within an Akt consensus phosphorylation motif (RxRxxS/T), and that this methylation blocks Akt-mediated phosphorylation of the transcription factors. These findings led us to hypothesize that the functional crosstalk between arginine methylation and phosphorylation could be extended to other Akt target proteins as well as Forkhead box O proteins. Here we identify BCL-2 antagonist of cell death (BAD) as an additional substrate for PRMT1 among several Akt target proteins. We show that PRMT1 specifically binds and methylates BAD at Arg-94 and Arg-96, both of which comprise the Akt consensus phosphorylation motif. Consistent with the hypothesis, PRMT1-mediated methylation of these two arginine residues inhibits Akt-mediated phosphorylation of BAD at Ser-99 in vitro and in vivo. We also demonstrate that the complex formation of BAD with 14-3-3 proteins, which occurs subsequent to Akt-mediated phosphorylation, is negatively regulated by PRMT1. Furthermore, PRMT1 knockdown prevents mitochondrial localization of BAD and its binding to the antiapoptotic BCL-X(L) protein. BAD overexpression causes an increase in apoptosis with concomitant activation of caspase-3, whereas PRMT1 knockdown significantly suppresses these apoptotic processes. Taken together, our results add a new dimension to the complexity of posttranslational BAD regulation and provide evidence that arginine methylation within an Akt consensus phosphorylation motif functions as an inhibitory modification against Akt-dependent survival signaling. | | | 21444773
|
BTG1 regulates glucocorticoid receptor autoinduction in acute lymphoblastic leukemia.
van Galen JC, Kuiper RP, van Emst L, Levers M, Tijchon E, Scheijen B, Waanders E, van Reijmersdal SV, Gilissen C, van Kessel AG, Hoogerbrugge PM, van Leeuwen FN
Blood
115
4810-9. Epub 2010 Mar 30.
2010
Show Abstract
Resistance to glucocorticoids (GCs) is a major clinical problem in the treatment of acute lymphoblastic leukemia (ALL), but the underlying mechanisms are not well understood. Although mutations in the glucocorticoid receptor (GR) gene can give rise to therapy resistance in vitro, acquired somatic mutations in the GR are rarely encountered in patients. Here we report that the protein encoded by the BTG1 gene, which is frequently deleted in (pediatric) ALL, is a key determinant of GC responsiveness. Using RNA interference, we show that loss of BTG1 expression causes GC resistance both by decimating GR expression and by controlling GR-mediated transcription. Conversely, reexpression of BTG1 restores GC sensitivity by potentiating GC-induced GR expression, a phenomenon known as GR autoinduction. In addition, the arginine methyltransferase PRMT1, a BTG1-binding partner and transcriptional coactivator, is recruited to the GR gene promoter in a BTG1-dependent manner. These results implicate the BTG1/PRMT1 complex in GR-mediated gene expression and reveal that deregulation of a nuclear receptor coactivator complex can give rise to GC resistance. Further characterization of this complex as part of the GR regulatory circuitry could offer novel opportunities for improving the efficacy of GC-based therapies in ALL and other hematologic malignancies. | | | 20354172
|