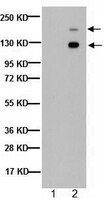
The sheddase ADAM10 is a potent modulator of prion disease.
Altmeppen, HC; Prox, J; Krasemann, S; Puig, B; Kruszewski, K; Dohler, F; Bernreuther, C; Hoxha, A; Linsenmeier, L; Sikorska, B; Liberski, PP; Bartsch, U; Saftig, P; Glatzel, M
eLife
4
2015
Show Abstract
The prion protein (PrP(C)) is highly expressed in the nervous system and critically involved in prion diseases where it misfolds into pathogenic PrP(Sc). Moreover, it has been suggested as a receptor mediating neurotoxicity in common neurodegenerative proteinopathies such as Alzheimer's disease. PrP(C) is shed at the plasma membrane by the metalloprotease ADAM10, yet the impact of this on prion disease remains enigmatic. Employing conditional knockout mice, we show that depletion of ADAM10 in forebrain neurons leads to posttranslational increase of PrP(C) levels. Upon prion infection of these mice, clinical, biochemical, and morphological data reveal that lack of ADAM10 significantly reduces incubation times and increases PrP(Sc) formation. In contrast, spatiotemporal analysis indicates that absence of shedding impairs spread of prion pathology. Our data support a dual role for ADAM10-mediated shedding and highlight the role of proteolytic processing in prion disease. | | | 25654651
 |
Exclusion of the unfolded protein response in light-induced retinal degeneration in the canine T4R RHO model of autosomal dominant retinitis pigmentosa.
Marsili, S; Genini, S; Sudharsan, R; Gingrich, J; Aguirre, GD; Beltran, WA
PloS one
10
e0115723
2015
Show Abstract
To examine the occurrence of endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) following acute light damage in the naturally-occurring canine model of RHO-adRP (T4R RHO dog).The left eyes of T4R RHO dogs were briefly light-exposed and retinas collected 3, 6 and 24 hours later. The contra-lateral eyes were shielded and used as controls. To evaluate the time course of cell death, histology and TUNEL assays were performed. Electron microscopy was used to examine ultrastructural alterations in photoreceptors at 15 min, 1 hour, and 6 hours after light exposure. Gene expression of markers of ER stress and UPR were assessed by RT-PCR, qRT-PCR and western blot at the 6 hour time-point. Calpain and caspase-3 activation were assessed at 1, 3 and 6 hours after exposure.A brief exposure to clinically-relevant levels of white light causes within minutes acute disruption of the rod outer segment disc membranes, followed by prominent ultrastructural alterations in the inner segments and the initiation of cell death by 6 hours. Activation of the PERK and IRE1 pathways, and downstream targets (BIP, CHOP) of the UPR was not observed. However increased transcription of caspase-12 and hsp70 occurred, as well as calpain activation, but not that of caspase-3.The UPR is not activated in the early phase of light-induced photoreceptor cell death in the T4R RHO model. Instead, disruption in rods of disc and plasma membranes within minutes after light exposure followed by increase in calpain activity and caspase-12 expression suggests a different mechanism of degeneration. | | | 25695253
|
Expression of Nampt in hippocampal and cortical excitatory neurons is critical for cognitive function.
Stein, LR; Wozniak, DF; Dearborn, JT; Kubota, S; Apte, RS; Izumi, Y; Zorumski, CF; Imai, S
The Journal of neuroscience : the official journal of the Society for Neuroscience
34
5800-15
2014
Show Abstract
Nicotinamide adenine dinucleotide (NAD(+)) is an enzyme cofactor or cosubstrate in many essential biological pathways. To date, the primary source of neuronal NAD(+) has been unclear. NAD(+) can be synthesized from several different precursors, among which nicotinamide is the substrate predominantly used in mammals. The rate-limiting step in the NAD(+) biosynthetic pathway from nicotinamide is performed by nicotinamide phosphoribosyltransferase (Nampt). Here, we tested the hypothesis that neurons use intracellular Nampt-mediated NAD(+) biosynthesis by generating and evaluating mice lacking Nampt in forebrain excitatory neurons (CaMKIIαNampt(-/-) mice). CaMKIIαNampt(-/-) mice showed hippocampal and cortical atrophy, astrogliosis, microgliosis, and abnormal CA1 dendritic morphology by 2-3 months of age. Importantly, these histological changes occurred with altered intrahippocampal connectivity and abnormal behavior; including hyperactivity, some defects in motor skills, memory impairment, and reduced anxiety, but in the absence of impaired sensory processes or long-term potentiation of the Schaffer collateral pathway. These results clearly demonstrate that forebrain excitatory neurons mainly use intracellular Nampt-mediated NAD(+) biosynthesis to mediate their survival and function. Studying this particular NAD(+) biosynthetic pathway in these neurons provides critical insight into their vulnerability to pathophysiological stimuli and the development of therapeutic and preventive interventions for their preservation. | | | 24760840
|
Ammonia mediates methamphetamine-induced increases in glutamate and excitotoxicity.
Halpin, LE; Northrop, NA; Yamamoto, BK
Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology
39
1031-8
2014
Show Abstract
Ammonia has been identified to have a significant role in the long-term damage to dopamine and serotonin terminals produced by methamphetamine (METH), but how ammonia contributes to this damage is unknown. Experiments were conducted to identify whether increases in brain ammonia affect METH-induced increases in glutamate and subsequent excitotoxicity. Increases in striatal glutamate were measured using in vivo microdialysis. To examine the role of ammonia in mediating changes in extracellular glutamate after METH exposure, lactulose was used to decrease plasma and brain ammonia. Lactulose is a non-absorbable disaccharide, which alters the intestinal lumen through multiple mechanisms that lead to the increased peripheral excretion of ammonia. METH caused a significant increase in extracellular glutamate that was prevented by lactulose. Lactulose had no effect on METH-induced hyperthermia. To determine if ammonia contributed to excitotoxicity, the effect of METH and lactulose treatment on calpain-mediated spectrin proteolysis was measured. METH significantly increased calpain-specific spectrin breakdown products, and this increase was prevented with lactulose treatment. To examine if ammonia-induced increases in extracellular glutamate were mediated by excitatory amino-acid transporters, the reverse dialysis of ammonia, the glutamate transporter inhibitor, DL-threo-β-benzyloxyaspartic acid (TBOA), or the combination of the two directly into the striatum of awake, freely moving rats was conducted. TBOA blocked the increases in extracellular glutamate produced by the reverse dialysis of ammonia. These findings demonstrate that ammonia mediates METH-induced increases in extracellular glutamate through an excitatory amino-acid transporter to cause excitotoxicity. | | | 24165886
|
Reduced migration of MLH1 deficient colon cancer cells depends on SPTAN1.
Hinrichsen, I; Ernst, BP; Nuber, F; Passmann, S; Schäfer, D; Steinke, V; Friedrichs, N; Plotz, G; Zeuzem, S; Brieger, A
Molecular cancer
13
11
2014
Show Abstract
Defects in the DNA mismatch repair (MMR) protein MLH1 are frequently observed in sporadic and hereditary colorectal cancers (CRC). Affected tumors generate much less metastatic potential than the MLH1 proficient forms. Although MLH1 has been shown to be not only involved in postreplicative MMR but also in several MMR independent processes like cytoskeletal organization, the connection between MLH1 and metastasis remains unclear. We recently identified non-erythroid spectrin αII (SPTAN1), a scaffolding protein involved in cell adhesion and motility, to interact with MLH1. In the current study, the interaction of MLH1 and SPTAN1 and its potential consequences for CRC metastasis was evaluated.Nine cancer cell lines as well as fresh and paraffin embedded colon cancer tissue from 12 patients were used in gene expression studies of SPTAN1 and MLH1. Co-expression of SPTAN1 and MLH1 was analyzed by siRNA knock down of MLH1 in HeLa, HEK293, MLH1 positive HCT116, SW480 and LoVo cells. Effects on cellular motility were determined in MLH1 deficient HCT116 and MLH1 deficient HEK293T compared to their MLH1 proficient sister cells, respectively.MLH1 deficiency is clearly associated with SPTAN1 reduction. Moreover, siRNA knock down of MLH1 decreased the mRNA level of SPTAN1 in HeLa, HEK293 as well as in MLH1 positive HCT116 cells, which indicates a co-expression of SPTAN1 by MLH1. In addition, cellular motility of MLH1 deficient HCT116 and MLH1 deficient HEK293T cells was impaired compared to the MLH1 proficient sister clones. Consequently, overexpression of SPTAN1 increased migration of MLH1 deficient cells while knock down of SPTAN1 decreased cellular mobility of MLH1 proficient cells, indicating SPTAN1-dependent migration ability.These data suggest that SPTAN1 levels decreased in concordance with MLH1 reduction and impaired cellular mobility in MLH1 deficient colon cancer cells. Therefore, aggressiveness of MLH1-positive CRC might be related to SPTAN1. | | | 24456667
|
EspC promotes epithelial cell detachment by enteropathogenic Escherichia coli via sequential cleavages of a cytoskeletal protein and then focal adhesion proteins.
Navarro-Garcia, F; Serapio-Palacios, A; Vidal, JE; Salazar, MI; Tapia-Pastrana, G
Infection and immunity
82
2255-65
2014
Show Abstract
EspC is a non-locus of enterocyte effacement (LEE)-encoded autotransporter produced by enteropathogenic Escherichia coli (EPEC) that is secreted to the extracellular milieu by a type V secretion system and then translocated into epithelial cells by the type III secretion system. Here, we show that this efficient EspC delivery into the cell leads to a cytopathic effect characterized by cell rounding and cell detachment. Thus, EspC is the main protein involved in epithelial cell cytotoxicity detected during EPEC adhesion and pedestal formation assays. The cell detachment phenotype is triggered by cytoskeletal and focal adhesion disruption. EspC-producing EPEC is able to cleave fodrin, paxillin, and focal adhesion kinase (FAK), but these effects are not observed when cells are infected with an espC isogenic mutant. Recovery of these phenotypes by complementing the mutant with the espC gene but not with the espC gene mutated in the serine protease motif highlights the role of the protease activity of EspC in the cell detachment phenotype. In vitro assays using purified proteins showed that EspC, but not EspC with an S256I substitution [EspCS256I], directly cleaves these cytoskeletal and focal adhesion proteins. Kinetics of protein degradation indicated that EspC-producing EPEC first cleaves fodrin (within the 11th and 9th repetitive units at the Q1219 and D938 residues, respectively), and this event sequentially triggers paxillin degradation, FAK dephosphorylation, and FAK degradation. Thus, cytoskeletal and focal adhesion protein cleavage leads to the cell rounding and cell detachment promoted by EspC-producing EPEC. | | | 24643541
|
Systemic and Cerebral Vascular Endothelial Growth Factor Levels Increase in Murine Cerebral Malaria along with Increased Calpain and Caspase Activity and Can be Reduced by Erythropoietin Treatment.
Hempel, C; Hoyer, N; Kildemoes, A; Jendresen, CB; Kurtzhals, JA
Frontiers in immunology
5
291
2014
Show Abstract
The pathogenesis of cerebral malaria (CM) includes compromised microvascular perfusion, increased inflammation, cytoadhesion, and endothelial activation. These events cause blood-brain barrier disruption and neuropathology and associations with the vascular endothelial growth factor (VEGF) signaling pathway have been shown. We studied this pathway in mice infected with Plasmodium berghei ANKA causing murine CM with or without the use of erythropoietin (EPO) as adjunct therapy. ELISA and western blotting was used for quantification of VEGF and relevant proteins in brain and plasma. CM increased levels of VEGF in brain and plasma and decreased plasma levels of soluble VEGF receptor 2. EPO treatment normalized VEGF receptor 2 levels and reduced brain VEGF levels. Hypoxia-inducible factor (HIF)-1α was significantly upregulated whereas cerebral HIF-2α and EPO levels remained unchanged. Furthermore, we noticed increased caspase-3 and calpain activity in terminally ill mice, as measured by protease-specific cleavage of α-spectrin and p35. In conclusion, we detected increased cerebral and systemic VEGF as well as HIF-1α, which in the brain were reduced to normal in EPO-treated mice. Also caspase and calpain activity was reduced markedly in EPO-treated mice. | | | 24995009
|
Inhibition of calpain-regulated p35/cdk5 plays a central role in sildenafil-induced protection against chemical hypoxia produced by malonate.
Barros-Miñones, L; Martín-de-Saavedra, D; Perez-Alvarez, S; Orejana, L; Suquía, V; Goñi-Allo, B; Hervias, I; López, MG; Jordan, J; Aguirre, N; Puerta, E
Biochimica et biophysica acta
1832
705-17
2013
Show Abstract
Phosphodiesterase 5 (PDE5) inhibitors have recently been reported to exert beneficial effects against ischemia-reperfusion injury in several organs but their neuroprotective effects in brain stroke models are scarce. The present study was undertaken to assess the effects of sildenafil against cell death caused by intrastriatal injection of malonate, an inhibitor of succinate dehydrogenase; which produces both energy depletion and lesions similar to those seen in cerebral ischemia. Our data demonstrate that sildenafil (1.5mg/kg by mouth (p.o.)), given 30min before malonate (1.5μmol/2μL), significantly decreased the lesion volume caused by this toxin. This protective effect can be probably related to the inhibition of excitotoxic pathways. Thus, malonate induced the activation of the calcium-dependent protease, calpain and the cyclin-dependent kinase 5, cdk5; which resulted in the hyperphosphorylation of tau and the cleavage of the protective transcription factor, myocyte enhancer factor 2, MEF2. All these effects were also significantly reduced by sildenafil pre-treatment, suggesting that sildenafil protects against malonate-induced cell death through the regulation of the calpain/p25/cdk5 signaling pathway. Similar findings were obtained using inhibitors of calpain or cdk5, further supporting our contention. Sildenafil also increased MEF2 phosphorylation and Bcl-2/Bax and Bcl-xL/Bax ratios, effects that might as well contribute to prevent cell death. Finally, sildenafil neuroprotection was extended not only to rat hippocampal slices subjected to oxygen and glucose deprivation when added at the time of reoxygenation, but also, in vivo when administered after malonate injection. Thus, the therapeutic window for sildenafil against malonate-induced hypoxia was set at 3h. | | | 23415811
|
Prevention of JNK phosphorylation as a mechanism for rosiglitazone in neuroprotection after transient cerebral ischemia: activation of dual specificity phosphatase.
Okami, N; Narasimhan, P; Yoshioka, H; Sakata, H; Kim, GS; Jung, JE; Maier, CM; Chan, PH
Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism
33
106-14
2013
Show Abstract
Rosiglitazone, a synthetic peroxisome proliferator-activated receptor-γ (PPARγ) agonist, prevents cell death after cerebral ischemia in animal models, but the underlying mechanism has not been clarified. In this study, we examined how rosiglitazone protects neurons against ischemia. Mice treated with rosiglitazone were subjected to 60 minutes of focal ischemia followed by reperfusion. Rosiglitazone reduced infarct volume after ischemia and reperfusion. We show that this neuroprotective effect was reversed with a PPARγ antagonist. Western blot analysis showed a significant increase in expression of phosphorylated stress-activated protein kinases (c-Jun N-terminal kinase (JNK) and p38) in ischemic brain tissue. Rosiglitazone blocked this increase. Furthermore, we observed that rosiglitazone increased expression of the dual-specificity phosphatase 8 (DUSP8) protein and messenger RNA in ischemic brain tissue. Dual-specificity phosphatase 8 is a mitogen-activated protein kinase phosphatase that can dephosphorylate JNK and p38. Another key finding of the present study was that knockdown of DUSP8 in primary cultured cortical neurons that were subjected to oxygen-glucose deprivation diminished rosiglitazone's effect on downregulation of JNK phosphorylation. Thus, rosiglitazone's neuroprotective effect after ischemia is mediated by blocking JNK phosphorylation induced by ischemia via DUSP8 upregulation. | Western Blotting | | 23032483
|
Negative regulation of 26S proteasome stability via calpain-mediated cleavage of Rpn10 subunit upon mitochondrial dysfunction in neurons.
Huang, Q; Wang, H; Perry, SW; Figueiredo-Pereira, ME
The Journal of biological chemistry
288
12161-74
2013
Show Abstract
Proteasomal and mitochondrial dysfunctions are implicated in chronic neurodegenerative diseases. To investigate the impact of mitochondrial impairment on the proteasome, we treated rat cerebral cortical neurons with oligomycin, antimycin, or rotenone, which inhibit different elements of the electron transport chain. Firstly, we observed a reduction in ubiquitinated proteins and E1 activity. Secondly, we established that 26S proteasomes are disassembled with a decline in activity. Thirdly, we show, to our knowledge for the first time, that calpain activation triggers the selective processing of the 26S proteasome subunit Rpn10. Other proteasome subunits tested were not affected. Calpain also cleaved caspase 3 to an inactive fragment, thus preventing apoptosis that is an energy-dependent cell death pathway. In addition, calpain cleaved the microtubule-associated protein Tau, a major component of neurofibrillary tangles in Alzheimer disease and other tauopathies. Fourthly, we detected a rise in 20S proteasome levels and activity. Finally, we show that both acute (16 h) and long term (up to 7 days) mitochondrial impairment led to down-regulation of ubiquitinated-proteins, 26S proteasome disassembly, and a rise in 20S proteasomes. We postulate that upon mitochondrial dysfunction, ATP depletion and calpain activation contribute to the demise of protein turnover by the ubiquitin/proteasome pathway. The concomitant rise in 20S proteasomes, which seem to degrade proteins in an unregulated and energy-independent manner, in the short term may carry out the turnover of randomly unfolded oxidized proteins. However, if chronic, it could lead to neurodegeneration as regulated protein degradation by the ubiquitin/proteasome pathway is essential for neuronal survival. | | | 23508964
|