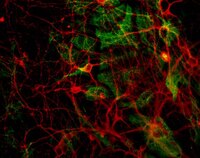
Huntington disease iPSCs show early molecular changes in intracellular signaling, the expression of oxidative stress proteins and the p53 pathway.
Szlachcic, WJ; Switonski, PM; Krzyzosiak, WJ; Figlerowicz, M; Figiel, M
Disease models & mechanisms
8
1047-57
2015
Show Abstract
Huntington disease (HD) is a brain disorder characterized by the late onset of motor and cognitive symptoms, even though the neurons in the brain begin to suffer dysfunction and degeneration long before symptoms appear. There is currently no cure. Several molecular and developmental effects of HD have been identified using neural stem cells (NSCs) and differentiated cells, such as neurons and astrocytes. Still, little is known regarding the molecular pathogenesis of HD in pluripotent cells, such as embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). Therefore, we examined putative signaling pathways and processes involved in HD pathogenesis in pluripotent cells. We tested naïve mouse HD YAC128 iPSCs and two types of human HD iPSC that were generated from HD and juvenile-HD patients. Surprisingly, we found that a number of changes affecting cellular processes in HD were also present in undifferentiated pluripotent HD iPSCs, including the dysregulation of the MAPK and Wnt signaling pathways and the dysregulation of the expression of genes related to oxidative stress, such as Sod1. Interestingly, a common protein interactor of the huntingtin protein and the proteins in the above pathways is p53, and the expression of p53 was dysregulated in HD YAC128 iPSCs and human HD iPSCs. In summary, our findings demonstrate that multiple molecular pathways that are characteristically dysregulated in HD are already altered in undifferentiated pluripotent cells and that the pathogenesis of HD might begin during the early stages of life. | Western Blotting | | 26092128
 |
A novel method to derive and expand mice neural stem cells efficiently without neuro-sphere formation.
Ma, ZZ; Fan, L; Huang, JL; Pan, XJ
International journal of clinical and experimental medicine
8
12834-41
2015
Show Abstract
Neural stem cells (NSCs) are multi-potent stem cells able to self-renew and generate immature and differentiated cell populations by asymmetric division. The NSCs are of considerable interest for cell replacement in neuro-degenerative diseases. NSCs are usually identified and expanded by their ability to generate free-floating aggregates termed neurospheres. However, neurospheres are not a pure population of NSCs with as little as 1% population in primary spheres. Neurospheres also contain neurons, astrocytes and oligodendrocytes. The heterogeneity of these cells may hinder their repopulation potential when used in cell transplantation. Furthermore, to obtain 1 million NSCs by the neurosphere protocol usually takes one month, which is inconvenient for future clinical trials. In this study, we tried to derive the NSCs from mice embryo neuroepithelium without neurosphere formation. Three different protocols were compared. We generated a direct and efficient NSCs generation, expanding and freezing protocol. This protocol can provide sufficient amount of the NSCs from first a few passages for cell transplantation. | | | 26550198
|
In vivo reprogrammed pluripotent stem cells from teratomas share analogous properties with their in vitro counterparts.
Choi, HW; Kim, JS; Hong, YJ; Song, H; Seo, HG; Do, JT
Scientific reports
5
13559
2015
Show Abstract
Recently, induced pluripotent stem cells (iPSCs) have been generated in vivo from reprogrammable mice. These in vivo iPSCs display features of totipotency, i.e., they differentiate into the trophoblast lineage, as well as all 3 germ layers. Here, we developed a new reprogrammable mouse model carrying an Oct4-GFP reporter gene to facilitate the detection of reprogrammed pluripotent stem cells. Without doxycycline administration, some of the reprogrammable mice developed aggressively growing teratomas that contained Oct4-GFP(+) cells. These teratoma-derived in vivo PSCs were morphologically indistinguishable from ESCs, expressed pluripotency markers, and could differentiate into tissues of all 3 germ layers. However, these in vivo reprogrammed PSCs were more similar to in vitro iPSCs than ESCs and did not contribute to the trophectoderm of the blastocysts after aggregation with 8-cell embryos. Therefore, the ability to differentiate into the trophoblast lineage might not be a unique characteristic of in vivo iPSCs. | | | 26315499
|
PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia.
Radford, H; Moreno, JA; Verity, N; Halliday, M; Mallucci, GR
Acta neuropathologica
130
633-42
2015
Show Abstract
The PERK-eIF2α branch of the Unfolded Protein Response (UPR) mediates the transient shutdown of translation in response to rising levels of misfolded proteins in the endoplasmic reticulum. PERK and eIF2α activation are increasingly recognised in postmortem analyses of patients with neurodegenerative disorders, including Alzheimer's disease, the tauopathies and prion disorders. These are all characterised by the accumulation of misfolded disease-specific proteins in the brain in association with specific patterns of neuronal loss, but the role of UPR activation in their pathogenesis is unclear. In prion-diseased mice, overactivation of PERK-P/eIF2α-P signalling results in the sustained reduction in global protein synthesis, leading to synaptic failure, neuronal loss and clinical disease. Critically, restoring vital neuronal protein synthesis rates by inhibiting the PERK-eIF2α pathway, both genetically and pharmacologically, prevents prion neurodegeneration downstream of misfolded prion protein accumulation. Here we show that PERK-eIF2α-mediated translational failure is a key process leading to neuronal loss in a mouse model of frontotemporal dementia, where the misfolded protein is a form of mutant tau. rTg4510 mice, which overexpress the P301L tau mutation, show dysregulated PERK signalling and sustained repression of protein synthesis by 6 months of age, associated with onset of neurodegeneration. Treatment with the PERK inhibitor, GSK2606414, from this time point in mutant tau-expressing mice restores protein synthesis rates, protecting against further neuronal loss, reducing brain atrophy and abrogating the appearance of clinical signs. Further, we show that PERK-eIF2α activation also contributes to the pathological phosphorylation of tau in rTg4510 mice, and that levels of phospho-tau are lowered by PERK inhibitor treatment, providing a second mechanism of protection. The data support UPR-mediated translational failure as a generic pathogenic mechanism in protein-misfolding disorders, including tauopathies, that can be successfully targeted for prevention of neurodegeneration. | | | 26450683
|
Genome-wide characterisation of Foxa1 binding sites reveals several mechanisms for regulating neuronal differentiation in midbrain dopamine cells.
Metzakopian, E; Bouhali, K; Alvarez-Saavedra, M; Whitsett, JA; Picketts, DJ; Ang, SL
Development (Cambridge, England)
142
1315-24
2015
Show Abstract
Midbrain dopamine neuronal progenitors develop into heterogeneous subgroups of neurons, such as substantia nigra pars compacta, ventral tegmental area and retrorubal field, that regulate motor control, motivated and addictive behaviours. The development of midbrain dopamine neurons has been extensively studied, and these studies indicate that complex cross-regulatory interactions between extrinsic and intrinsic molecules regulate a precise temporal and spatial programme of neurogenesis in midbrain dopamine progenitors. To elucidate direct molecular interactions between multiple regulatory factors during neuronal differentiation in mice, we characterised genome-wide binding sites of the forkhead/winged helix transcription factor Foxa1, which functions redundantly with Foxa2 to regulate the differentiation of mDA neurons. Interestingly, our studies identified a rostral brain floor plate Neurog2 enhancer that requires direct input from Otx2, Foxa1, Foxa2 and an E-box transcription factor for its transcriptional activity. Furthermore, the chromatin remodelling factor Smarca1 was shown to function downstream of Foxa1 and Foxa2 to regulate differentiation from immature to mature midbrain dopaminergic neurons. Our genome-wide Foxa1-bound cis-regulatory sequences from ChIP-Seq and Foxa1/2 candidate target genes from RNA-Seq analyses of embryonic midbrain dopamine cells also provide an excellent resource for probing mechanistic insights into gene regulatory networks involved in the differentiation of midbrain dopamine neurons. | | | 25804738
|
Properties of neurons derived from induced pluripotent stem cells of Gaucher disease type 2 patient fibroblasts: potential role in neuropathology.
Sun, Y; Florer, J; Mayhew, CN; Jia, Z; Zhao, Z; Xu, K; Ran, H; Liou, B; Zhang, W; Setchell, KD; Gu, J; Grabowski, GA
PloS one
10
e0118771
2015
Show Abstract
Gaucher disease (GD) is caused by insufficient activity of acid β-glucosidase (GCase) resulting from mutations in GBA1. To understand the pathogenesis of the neuronopathic GD, induced pluripotent stem cells (iPSCs) were generated from fibroblasts isolated from three GD type 2 (GD2) and 2 unaffected (normal and GD carrier) individuals. The iPSCs were converted to neural precursor cells (NPCs) which were further differentiated into neurons. Parental GD2 fibroblasts as well as iPSCs, NPCs, and neurons had similar degrees of GCase deficiency. Lipid analyses showed increases of glucosylsphingosine and glucosylceramide in the GD2 cells. In addition, GD2 neurons showed increased α-synuclein protein compared to control neurons. Whole cell patch-clamping of the GD2 and control iPSCs-derived neurons demonstrated excitation characteristics of neurons, but intriguingly, those from GD2 exhibited consistently less negative resting membrane potentials with various degree of reduction in action potential amplitudes, sodium and potassium currents. Culture of control neurons in the presence of the GCase inhibitor (conduritol B epoxide) recapitulated these findings, providing a functional link between decreased GCase activity in GD and abnormal neuronal electrophysiological properties. To our knowledge, this study is first to report abnormal electrophysiological properties in GD2 iPSC-derived neurons that may underlie the neuropathic phenotype in Gaucher disease. | Immunofluorescence | | 25822147
|
Macromolecular organization and fine structure of the human basilar membrane - RELEVANCE for cochlear implantation.
Liu, W; Atturo, F; Aldaya, R; Santi, P; Cureoglu, S; Obwegeser, S; Glueckert, R; Pfaller, K; Schrott-Fischer, A; Rask-Andersen, H
Cell and tissue research
360
245-62
2015
Show Abstract
Cochlear micromechanics and frequency tuning depend on the macromolecular organization of the basilar membrane (BM), which is still unclear in man. Novel techniques in cochlear implantation (CI) motivate further analyses of the BM.Normal cochleae from patients undergoing removal of life-threatening petro-clival meningioma and an autopsy specimen from a normal human were used. Laser-confocal microscopy, high resolution scanning (SEM) and transmission electron microscopy (TEM) were carried out in combination. In addition, one human temporal bone was decellularized and investigated by SEM.The human BM consisted in four separate layers: (1) epithelial basement membrane positive for laminin-β2 and collagen IV, (2) BM "proper" composed of radial fibers expressing collagen II and XI, (3) layer of collagen IV and (4) tympanic covering layer (TCL) expressing collagen IV, fibronectin and integrin. BM thickness varied both radially and longitudinally (mean 0.55-1.16 μm). BM was thinnest near the OHC region and laterally.There are several important similarities and differences between the morphology of the BM in humans and animals. Unlike in animals, it does not contain a distinct pars tecta (arcuate) and pectinata. Its width increases and thickness decreases as it travels apically in the cochlea. Findings show that the human BM is thinnest and probably most vibration-sensitive at the outer pillar feet/Deiter cells at the OHCs. The inner pillar and IHCs seem situated on a fairly rigid part of the BM. The gradient design of the BM suggests that its vulnerability increases apical wards when performing hearing preservation CI surgery. | | | 25663274
|
Glycaemic regulation and insulin secretion are abnormal in cystic fibrosis pigs despite sparing of islet cell mass.
Uc, A; Olivier, AK; Griffin, MA; Meyerholz, DK; Yao, J; Abu-El-Haija, M; Buchanan, KM; Vanegas Calderón, OG; Abu-El-Haija, M; Pezzulo, AA; Reznikov, LR; Hoegger, MJ; Rector, MV; Ostedgaard, LS; Taft, PJ; Gansemer, ND; Ludwig, PS; Hornick, EE; Stoltz, DA; Ode, KL; Welsh, MJ; Engelhardt, JF; Norris, AW
Clinical science (London, England : 1979)
128
131-42
2015
Show Abstract
Diabetes is a common and significant co-morbidity in cystic fibrosis (CF). The pathogenesis of cystic fibrosis related diabetes (CFRD) is incompletely understood. Because exocrine pancreatic disease is similar between humans and pigs with CF, the CF pig model has the potential to contribute significantly to the understanding of CFRD pathogenesis. We determined the structure of the endocrine pancreas in fetal, newborn and older CF and non-CF pigs and assessed endocrine pancreas function by intravenous glucose tolerance test (IV-GTT). In fetal pigs, pancreatic insulin and glucagon density was similar between CF and non-CF. In newborn and older pigs, the insulin and glucagon density was unchanged between CF and non-CF per total pancreatic area, but increased per remnant lobular tissue in CF reflecting exocrine pancreatic loss. Although fasting glucose levels were not different between CF and non-CF newborns, CF newborns demonstrated impaired glucose tolerance and increased glucose area under the curve during IV-GTT. Second phase insulin secretion responsiveness was impaired in CF newborn pigs and significantly lower than that observed in non-CF newborns. Older CF pigs had elevated random blood glucose levels compared with non-CF. In summary, glycaemic abnormalities and insulin secretion defects were present in newborn CF pigs and spontaneous hyperglycaemia developed over time. Functional changes in CF pig pancreas were not associated with a decline in islet cell mass. Our results suggest that functional islet abnormalities, independent of structural islet loss, contribute to the early pathogenesis of CFRD. | | | 25142104
|
The pre- and post-somatic segments of the human type I spiral ganglion neurons--structural and functional considerations related to cochlear implantation.
Liu, W; Edin, F; Atturo, F; Rieger, G; Löwenheim, H; Senn, P; Blumer, M; Schrott-Fischer, A; Rask-Andersen, H; Glueckert, R
Neuroscience
284
470-82
2015
Show Abstract
Human auditory nerve afferents consist of two separate systems; one is represented by the large type I cells innervating the inner hair cells and the other one by the small type II cells innervating the outer hair cells. Type I spiral ganglion neurons (SGNs) constitute 96% of the afferent nerve population and, in contrast to other mammals, their soma and pre- and post-somatic segments are unmyelinated. Type II nerve soma and fibers are unmyelinated. Histopathology and clinical experience imply that human SGNs can persist electrically excitable without dendrites, thus lacking connection to the organ of Corti. The biological background to this phenomenon remains elusive. We analyzed the pre- and post-somatic segments of the type I human SGNs using immunohistochemistry and transmission electron microscopy (TEM) in normal and pathological conditions. These segments were found surrounded by non-myelinated Schwann cells (NMSCs) showing strong intracellular expression of laminin-β2/collagen IV. These cells also bordered the perikaryal entry zone and disclosed surface rugosities outlined by a folded basement membrane (BM) expressing laminin-β2 and collagen IV. It is presumed that human large SGNs are demarcated by three cell categories: (a) myelinated Schwann cells, (b) NMSCs and (c) satellite glial cells (SGCs). Their BMs express laminin-β2/collagen IV and reaches the BM of the sensory epithelium at the habenula perforata. We speculate that the NMSCs protect SGNs from further degeneration following dendrite loss. It may give further explanation why SGNs can persist as electrically excitable monopolar cells even after long-time deafness, a blessing for the deaf treated with cochlear implantation. | Immunohistochemistry | | 25316409
|
Astroblastoma: beside being a tumor entity, an occasional phenotype of astrocytic gliomas?
Mellai, M; Piazzi, A; Casalone, C; Grifoni, S; Melcarne, A; Annovazzi, L; Cassoni, P; Denysenko, T; Valentini, MC; Cistaro, A; Schiffer, D
OncoTargets and therapy
8
451-60
2015
Show Abstract
The diagnosis of astroblastoma is based on a typical histological aspect with perivascular distribution of cells sending cytoplasmic extensions to the vessels and vascular hyalinization. These criteria are useful for standardizing the identification of the tumor, but, in spite of this, there are discrepancies in the literature concerning the age distribution and the benign or malignant nature of the tumor. Three cases are discussed in this study: Case 1 was a typical high-grade astroblastoma; Case 2 was an oligodendroglioma at the first intervention and an oligoastrocytoma at the second intervention with typical perivascular arrangements in the astrocytic component; Case 3 was a gemistocytic glioma with malignant features and typical perivascular arrangements. Genetic analysis showed genetic alterations that are typical of gliomas of all malignancy grades. Using the neurosphere assay, neurospheres and adherent cells were found to have developed in Case 1, while adherent cells only developed in Case 2, in line with the stemness potential of the tumors. The cases are discussed in relation to their diagnostic assessment as astroblastoma, and it is hypothesized that the typical perivascular distribution of cells may not indicate a separate and unique tumor entity, but may be a peculiarity that can be acquired by astrocytic gliomas when an unknown cause from the tumor microenvironment influences the relationship between vessels and tumor cells. | Immunofluorescence | | 25737639
|