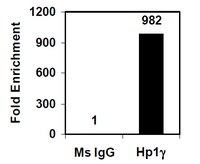
A functional CNVR_3425.1 damping lincRNA FENDRR increases lifetime risk of lung cancer and COPD in Chinese.
Yang, L; Wu, D; Chen, J; Chen, J; Qiu, F; Li, Y; Liu, L; Cao, Y; Yang, B; Zhou, Y; Lu, J
Carcinogenesis
39
347-359
2018
Show Abstract
Genomic imbalance referring to somatic variation in chromosome copies represents the most frequent event in tumorigenesis. Germline copy number variations (gCNVs) overlapping regions of genomic imbalance harbor similar structural characteristics and thus influence tumor susceptibility. We aimed to test effects of such gCNVs on the risk of lung cancer and chronic obstructive pulmonary disease (COPD). Genomic imbalance of lung cancer was determined by the array comparative genomic hybridization (aCGH), and common gCNVs at these imbalance regions were genotyped in lung cancer-based and COPD-based retrospective studies. Functional assays were conducted to assess function of promising CNVs. A total of 115 genomic imbalances were discovered occurring at a frequency of more than 25%. The CNVR_3425.1, overlapping the chr16q24.1 with genomic imbalance, was significantly associated with increased risks of lung cancer (OR = 1.76; 95% CI = 1.46-2.11) and COPD (OR = 1.98; 95% CI = 1.57-2.51). The increase copy of CNVR_3425.1 forms a new additional truncated FOXF1 adjacent non-coding developmental regulatory RNA (FENDRR) sequences comparing the gene promoter and perturbs the transcriptional factors (TFs) binding to the original FENDRR promoter and further downregulates FENDRR, a long intergenic non-coding RNA (lincRNA) that functions to inhibit lung cancer by affecting expressions of an abundant number of genes, including the tumor suppressor FOXF1. FENDRR can upregulate FOXF1 by competitively binding to miR-424. The TFs early growth response 1 (EGR1) and transcription factor AP-2 alpha (TFAP2A) were further found to involve the CNVR_3425.1-mediated FENDRR dysregulation. These findings suggested the CNVR_3425.1 to be a possibly predictive biomarker for the risk of lung cancer and COPD, and targeted molecular therapy pertaining to FENDRR upregulation may be a valuable pathway to fight two diseases. | 29293945
 |
Long noncoding RNA BLACAT2 promotes bladder cancer-associated lymphangiogenesis and lymphatic metastasis.
He, W; Zhong, G; Jiang, N; Wang, B; Fan, X; Chen, C; Chen, X; Huang, J; Lin, T
J Clin Invest
128
861-875
2018
Show Abstract
The prognosis for bladder cancer patients with lymph node (LN) metastasis is dismal and only minimally improved by current treatment modalities. Elucidation of the molecular mechanisms that underlie LN metastasis may provide clinical therapeutic strategies for LN-metastatic bladder cancer. Here, we report that a long noncoding RNA LINC00958, which we have termed bladder cancer-associated transcript 2 (BLACAT2), was markedly upregulated in LN-metastatic bladder cancer and correlated with LN metastasis. Overexpression of BLACAT2 promoted bladder cancer-associated lymphangiogenesis and lymphatic metastasis in both cultured bladder cancer cell lines and mouse models. Furthermore, we demonstrate that BLACAT2 epigenetically upregulated VEGF-C expression by directly associating with WDR5, a core subunit of human H3K4 methyltransferase complexes. Importantly, administration of an anti-VEGF-C antibody inhibited LN metastasis in BLACAT2-overexpressing bladder cancer. Taken together, these findings uncover a molecular mechanism in the lymphatic metastasis of bladder cancer and indicate that BLACAT2 may represent a target for clinical intervention in LN-metastatic bladder cancer. | 29355840
|
Ehrlichia chaffeensis TRP120 Effector Targets and Recruits Host Polycomb Group Proteins for Degradation To Promote Intracellular Infection.
Mitra, S; Dunphy, PS; Das, S; Zhu, B; Luo, T; McBride, JW
Infect Immun
86
2018
Show Abstract
Ehrlichia chaffeensis has a group of well-characterized type I secreted tandem repeat protein (TRP) effectors that have moonlighting capabilities. TRPs modulate various cellular processes, reprogram host gene transcription as nucleomodulins, function as ubiquitin ligases, and directly activate conserved host cell signaling pathways to promote E. chaffeensis infection. One TRP-interacting host target is polycomb group ring finger protein 5 (PCGF5), a member of the polycomb group (PcG) protein family and a component of the polycomb repressive complex 1 (PRC1). The current study demonstrates that during early infection, PCGF5 strongly colocalizes with TRP120 in the nucleus and later dramatically redistributes to the ehrlichial vacuole along with other PCGF isoforms. Ectopic expression and immunoprecipitation of TRP120 confirmed the interaction of TRP120 with multiple different PCGF isoforms. At 48 h postinfection, a dramatic redistribution of PCGF isoforms from the nucleus to the ehrlichial vacuole was observed, which also temporally coincided with proteasomal degradation of PCGF isoforms and TRP120 expression on the vacuole. A decrease in PRC1-mediated repressive chromatin mark and an altered transcriptional activity in PRC1-associated Hox genes primarily from HOXB and HOXC clusters were observed along with the degradation of PCGF isoforms, suggesting disruption of the PRC1 in E. chaffeensis-infected cells. Notably, small interfering RNA (siRNA)-mediated knockdown of PCGF isoforms resulted in significantly increased E. chaffeensis infection. This study demonstrates a novel strategy in which E. chaffeensis manipulates PRC complexes through interactions between TRP120 and PCGF isoforms to promote infection. | 29358333
|
Histone H3.3K27M Mobilizes Multiple Cancer/Testis (CT) Antigens in Pediatric Glioma.
Deng, H; Zeng, J; Zhang, T; Gong, L; Zhang, H; Cheung, E; Jones, C; Li, G
Mol Cancer Res
16
623-633
2018
Show Abstract
Lysine to methionine mutations at position 27 (K27M) in the histone H3 (H3.3 and H3.1) are highly prevalent in pediatric high-grade gliomas (HGG) that arise in the midline of the central nervous system. H3K27M perturbs the activity of polycomb repressor complex 2 and correlates with DNA hypomethylation; however, the pathways whereby H3K27M drives the development of pediatric HGG remain poorly understood. To understand the mechanism of pediatric HGG development driven by H3.3K27M and discover potential therapeutic targets or biomarkers, we established pediatric glioma cell model systems harboring H3.3K27M and performed microarray analysis. H3.3K27M caused the upregulation of multiple cancer/testis (CT) antigens, such as ADAMTS1, ADAM23, SPANXA1, SPANXB1/2, IL13RA2, VCY, and VCX3A, in pediatric glioma cells. Chromatin immunoprecipitation analysis from H3.3K27M cells revealed decreased H3K27me3 levels and increased H3K4me3 levels on the VCX3A promoter. Knockdown of VCX3A by siRNA significantly inhibited the growth of pediatric glioma cells harboring H3.3K27M. Overexpression of VCX3A/B genes stimulated the expression of several HLA genes, including HLA-A, HLA-B, HLA-E, HLA-F, and HLA-G The expression of VCX3A in pediatric HGG was confirmed using a tissue microarray. Gene set enrichment analysis revealed that CT antigens are enriched in pediatric HGG clinical specimens with H3.3K27M, with the upregulation of IL13RA2 contributing to the enrichment significantly. These results indicate that the upregulation of CT antigens, such as VCX3A and IL13RA2, correlates with pediatric gliomagenesis. Mol Cancer Res; 16(4); 623-33. ©2018 AACR. | 29453317
|
Suppression of the ERK-SRF axis facilitates somatic cell reprogramming.
Huh, S; Song, HR; Jeong, GR; Jang, H; Seo, NH; Lee, JH; Yi, JY; Lee, B; Choi, HW; Do, JT; Kim, JS; Lee, SH; Jung, JW; Lee, T; Shim, J; Han, MK; Lee, TH
Exp Mol Med
50
e448
2018
Show Abstract
The molecular mechanism underlying the initiation of somatic cell reprogramming into induced pluripotent stem cells (iPSCs) has not been well described. Thus, we generated single-cell-derived clones by using a combination of drug-inducible vectors encoding transcription factors (Oct4, Sox2, Klf4 and Myc) and a single-cell expansion strategy. This system achieved a high reprogramming efficiency after metabolic and epigenetic remodeling. Functional analyses of the cloned cells revealed that extracellular signal-regulated kinase (ERK) signaling was downregulated at an early stage of reprogramming and that its inhibition was a driving force for iPSC formation. Among the reprogramming factors, Myc predominantly induced ERK suppression. ERK inhibition upregulated the conversion of somatic cells into iPSCs through concomitant suppression of serum response factor (SRF). Conversely, SRF activation suppressed the reprogramming induced by ERK inhibition and negatively regulated embryonic pluripotency by inducing differentiation via upregulation of immediate early genes, such as c-Jun, c-Fos and EGR1. These data reveal that suppression of the ERK-SRF axis is an initial molecular event that facilitates iPSC formation and may be a useful surrogate marker for cellular reprogramming. | 29472703
|
COX-2/PGE2 Axis Regulates HIF2α Activity to Promote Hepatocellular Carcinoma Hypoxic Response and Reduce the Sensitivity of Sorafenib Treatment.
Dong, XF; Liu, TQ; Zhi, XT; Zou, J; Zhong, JT; Li, T; Mo, XL; Zhou, W; Guo, WW; Liu, X; Chen, YY; Li, MY; Zhong, XG; Han, YM; Wang, ZH; Dong, ZR
Clin Cancer Res
0
2018
Show Abstract
Purpose: Hypoxia-inducible factor-2α (HIF2α) is regarded as a preferential target for individualized hepatocellular carcinoma (HCC) treatment and sorafenib resistance. Our study aimed to identify the regulatory mechanisms of HIF2α activity under hypoxic conditions. We sought to determine whether the COX-2/PGE2 axis is involved in the regulatory mechanisms of HIF2α activity and of sorafenib resistance in hypoxic HCC cells.Experimental design: The cell viability, migration, and invasion abilities were measured to analyze the effects of HIF2α on hypoxic HCC cells. Both in vitro and in vivo HCC models were used to determine whether the COX-2/PGE2 axis is a driver of HIF2α level and activity, which then reduces the sensitivity of sorafenib treatment in hypoxic HCC cells.Results: Under hypoxic conditions, the COX-2/PGE2 axis effectively stabilized HIF2α and increased its level and activity via decreasing von Hippel-Lindau protein (p-VHL) level, and also enhanced HIF2α activity by promoting HIF2α nuclear translocation via MAPK pathway. The activation of HIF2α then led to the enhanced activation of VEGF, cyclin D1, and TGFα/EGFR pathway to mediate HCC development and reduce the sensitivity of sorafenib. More importantly, COX-2-specific inhibitors synergistically enhanced the antitumor activity of sorafenib treatment.Conclusions: Our data obtained demonstrate that the COX/PGE2 axis acts as a regulator of HIF2α expression and activity to promote HCC development and reduce sorafenib sensitivity by constitutively activating the TGFα/EGFR pathway. This study highlights the potential of COX-2-specific inhibitors for HCC treatment and particularly for enhancing the response to sorafenib treatment. Clin Cancer Res; 1-13. ©2018 AACR. | 29514844
|
Aberrant methylation-mediated silencing of lncRNA CTC-276P9.1 is associated with malignant progression of esophageal squamous cell carcinoma.
Guo, W; Liu, S; Dong, Z; Guo, Y; Ding, C; Shen, S; Liang, J; Shan, B
Clin Exp Metastasis
35
53-68
2018
Show Abstract
Downregulation and aberrant hypermethylation of long non-coding RNA CTC-276P9.1 have been detected in limited tumors. However, the distribution of methylated CpG sites and biological role of CTC-276P9.1 in esophageal squamous cell carcinoma (ESCC) progression and prognosis have not been fully clarified. The present study was to investigate the expression status and the distribution of methylated CpG sites within the three CpG islands of CTC-276P9.1, further to clarify its functional role and prognostic value in ESCC development and prognosis. Significant downregulation of CTC-276P9.1 was detected in esophageal cancer cells and ESCC tissues, and the expression of CTC-276P9.1 in ESCC tissues was associated with TNM stage, pathological differentiation, lymph node metastasis, and distant metastasis or recurrence. The expression level of CTC-276P9.1 in esophageal cancer cells was significantly reversed by treatment with 5-Aza-dC and TSA. The aberrant hypermethylation of the regions around the transcription start site was more tumor specific and associated with the expression levels of CTC-276P9.1. Moreover, histone modification may also participate in the regulation of CTC-276P9.1. Furthermore, over-expression of CTC-276P9.1 inhibited esophageal cancer cells proliferation and invasion in vitro, decreased the expression of proliferative markers and inhibited esophageal cancer cells invasion probably by regulating EMT. In addition, the dysregulation and hypermethylation of the regions around the transcription start site of CTC-276P9.1 were associated with poorer ESCC patients' survival. These findings suggest that CTC-276P9.1 may act as a tumor suppressor and may be employed as a new prognostic factor and therapeutic target for ESCC. | 29524086
|
BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency.
Sun, C; Yin, J; Fang, Y; Chen, J; Jeong, KJ; Chen, X; Vellano, CP; Ju, Z; Zhao, W; Zhang, D; Lu, Y; Meric-Bernstam, F; Yap, TA; Hattersley, M; O'Connor, MJ; Chen, H; Fawell, S; Lin, SY; Peng, G; Mills, GB
Cancer Cell
33
401-416.e8
2018
Show Abstract
Poly(ADP-ribose) polymerase inhibitors (PARPi) are selectively active in cells with homologous recombination (HR) deficiency (HRD) caused by mutations in BRCA1, BRCA2, and other pathway members. We sought small molecules that induce HRD in HR-competent cells to induce synthetic lethality with PARPi and extend the utility of PARPi. We demonstrated that inhibition of bromodomain containing 4 (BRD4) induced HRD and sensitized cells across multiple tumor lineages to PARPi regardless of BRCA1/2, TP53, RAS, or BRAF mutation status through depletion of the DNA double-stand break resection protein CtIP (C-terminal binding protein interacting protein). Importantly, BRD4 inhibitor (BRD4i) treatment reversed multiple mechanisms of resistance to PARPi. Furthermore, PARPi and BRD4i are synergistic in multiple in vivo models. | 29533782
|
Aberrant methylation-mediated downregulation of long noncoding RNA C5orf66-AS1 promotes the development of gastric cardia adenocarcinoma.
Guo, W; Lv, P; Liu, S; Xu, F; Guo, Y; Shen, S; Liang, J; Kuang, G; Dong, Z
Mol Carcinog
57
854-865
2018
Show Abstract
As a long non-coding RNA, C5orf66-AS1 is located at 5q31.1. Downregulation and aberrant hypermethylation of C5orf66-AS1 have been detected in a limited several tumors. However, the biological role and distribution of methylated CpG sites of C5orf66-AS1 in gastric cardia adenocarcinoma (GCA) development and prognosis are poorly clarified. The present study was to investigate the expression status and function of C5orf66-AS1 in GCA, and to detect the distribution of methylated CpG sites within the three CpG islands of the promoter and gene body of C5orf66-AS1, further to clarify its prognostic value in GCA patients. C5orf66-AS1 was significantly downregulated in GCA tissues and cell lines, and the expression level was associated with TNM stage, pathological differentiation, lymph node metastasis, and distant metastasis or recurrence. The expression level of C5orf66-AS1 was significantly increased in cancer cells after treated with 5-Aza-dC. Further methylation analysis demonstrated that the aberrant hypermethylation of the regions around the transcription start site of C5orf66-AS1 was more tumor specific and was associated with its expression. Moreover, Sp1 may upregulate C5orf66-AS1 expression and CpG sites hypermethylation within the binding sites may abrogate Sp1 binding. In addition, C5orf66-AS1 inhibited gastric cancer cell proliferation and invasion, and the dysregulation and hypermethylation of the regions around the transcription start site of C5orf66-AS1 were associated with poorer GCA patients' survival. These findings suggest that aberrant hypermethylation-mediated downregulation of C5orf66-AS1 may play important roles in GCA tumorigenesis and C5orf66-AS1 may serve as a potential prognostic marker in predicting GCA patients' survival. | 29566283
|
The effects of wild bitter gourd fruit extracts on ICAM-1 expression in pulmonary epithelial cells of C57BL/6J mice and microRNA-221/222 knockout mice: Involvement of the miR-221/-222/PI3K/AKT/NF-κB pathway.
Sung, HC; Liu, CW; Hsiao, CY; Lin, SR; Yu, IS; Lin, SW; Chiang, MH; Liang, CJ; Pu, CM; Chen, YC; Lin, MS; Chen, YL
Phytomedicine
42
90-99
2018
Show Abstract
The extracts from wild bitter gourd fruit (WBGE) were reported to possess numerous pharmacological activities. However, the anti-inflammatory effects of WBGE on human lung epithelial cells and the underlying mechanisms have not been determined.To evaluate the molecular basis of the effects of WBGE on intercellular adhesion molecule-1 (ICAM-1) expression in alveolar epithelial (A549) cells, C57BL/6 wild-type (WT) mice and microRNA (miR)-221/-222 knockout (KO) mice with or without tumor necrosis factor (TNF-α; 3 ng/ml) treatment.WT mice and miR-221/-222 KO mice were fed a control diet and divided into four groups (C: control mice; T: treated with TNF-α alone; WBGE/T: pretreated with WBGE and then stimulated with TNF-α; WBGE: treated with WBGE alone). The effects of WBGE on ICAM-1 expression and the related signals in A549 cells and mice with or without TNF-α treatment were examined by Western blot and immunofluorescent staining.WBGE significantly decreased the TNF-α-induced ICAM-1 expression in A549 cells through the inhibition of phosphoinositide 3-kinase (PI3K)/ protein kinase B (AKT)/ nuclear factor- kappa B (NF-κB)/ inhibitor of NF-κB (IκB) phosphorylation and decreased leukocyte adhesion. In addition, WBGE reduced endogenous ICAM-1 expression and upregulated miR-221/-222 expression. The overexpression of miR-222 decreased PI3K/AKT/NF-κB/IκB and ICAM-1 expression, which resulted in reducing monocyte adhesion. Moreover, WBGE reduced ICAM-1 expression in lung tissues of WT mice with or without TNF-α treatment and upregulated miR-221/222. WBGE did not affect the miR-221/-222 level and had little effect on ICAM-1 expression in miR-221/-222 KO mice.These results suggest that WBGE reduced ICAM-1 expression both under in vitro and in vivo conditions. The protective effects were mediated partly through the miR-221/-222/PI3K/AKT/NF-κB pathway. | 29655703
|