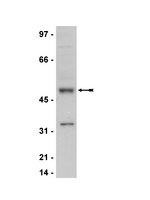
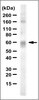
Cell cycle regulation in mouse heart during embryonic and postnatal stages.
Aiko Ikenishi,Hitomi Okayama,Noriko Iwamoto,Satoshi Yoshitome,Shoji Tane,Kazuomi Nakamura,Tetsuya Obayashi,Toshinori Hayashi,Takashi Takeuchi
Development, growth & differentiation
54
2012
Show Abstract
The regulation of cardiomyocyte proliferation is important for heart development and function. Proliferation levels of mouse cardiomyocytes are high during early embryogenesis and start to decrease at midgestation. Many cardiomyocytes undergo mitosis without cytokinesis, resulting in binucleated cardiomyocytes during early postnatal stages, following which the cell cycle arrests irreversibly. It remains unknown how the proliferation pattern is regulated, and how the irreversible cell cycle arrest occurs. To clarify the mechanisms, fundamental information about cell cycle regulators in cardiomyocytes and cell cycle patterns during embryonic and postnatal stages is necessary. Here, we show that the expression, complex formation, and activity of main cyclins and cyclin-dependent kinases (CDKs) changed in a synchronous manner during embryonic and postnatal stages. These levels decreased from midgestation to birth, and then showed one wave in which the peak was around postnatal day 5. Detailed analysis of the complexes suggested that CDK activities were inhibited before the protein levels decreased. Analysis of DNA content distribution patterns in mono- and binucleated cardiomyocytes after birth revealed changes in cell cycle distribution patterns and the transition from mono- to binucleated cells. These analyses indicated that the wave of cell cycle regulator expression or activities during postnatal stages mainly produced binucleated cells from mononucleated cells. The data obtained should provide a basis for the analysis of cell cycle regulation in cardiomyocytes during embryonic and postnatal stages. | | 22957921
 |
In Vitro Suppression of Growth of Murine WEHI-3 Leukemia Cells and in Vivo Promotion of Phagocytosis in a Leukemia Mice Model by Indole-3-carbinol.
Hsu-Feng Lu,Wei-Lin Tung,Jai-Sing Yang,Fang-Ming Huang,Ching-Sung Lee,Yi-Ping Huang,Wen-Yen Liao,Yung-Liang Chen,Jing-Gung Chung
Journal of agricultural and food chemistry
60
2012
Show Abstract
Indole-3-carbinol (I3C), a potential anticancer substance, can be found in cruciferous (cabbage family) vegetables, mainly cauliflower and Chinese cabbage. However, the bioactivity of I3C on the apoptotic effects of murine leukemia WEHI-3 cells and promotion of immune responses in leukemia mice model are unclear. In this study, we investigated the effect of I3C on cell-cycle arrest and apoptosis in vitro and immunomodulation in vivo. I3C decreased the viable WEHI-3 cells and caused morphological changes in a concentration- and time-dependent manner. I3C also led to G0/G1 phase arrest, decreased the levels of cyclin A, cyclin D, and CDK2, and increased the level of p21(WAF1/CIP1). Flow cytometric analyses further proved that I3C promoted ROS and intracellular Ca(2+) production and decreased the levels of ΔΨ(m) in WEHI-3 cells. Cells after exposure to I3C for 24 h showed DNA fragmentation and chromatin condensation. Comet assay also indicated that I3C induced DNA damage in examined cells. I3C increased the levels of cytochrome c, FADD, GADD153, GRP78, and caspase-12 as well as induced activities of caspase-3, -8, and -9. Moreover, I3C attenuated NF-κB DNA binding activity in I3C-treated WEHI-3 cells as shown by EMSA and Western blotting analyses. In the in vivo study, we examined the effects of I3C on WEHI-3 leukemia mice. Results showed that I3C increased the level of T cells and decreased the level of macrophages. I3C also reduced the weights of liver and spleen, and it promoted phagocytosis by macrophages as compared to the nontreated leukemia mice group. On the basis of our results, I3C affects murine leukemia WEHI-3 cells both in vitro and in vivo. | | 22775144
|
Cyclin E is recruited to the nuclear matrix during differentiation, but is not recruited in cancer cells.
Munkley, J; Copeland, NA; Moignard, V; Knight, JR; Greaves, E; Ramsbottom, SA; Pownall, ME; Southgate, J; Ainscough, JF; Coverley, D
Nucleic acids research
39
2671-7
2011
Show Abstract
Cyclin E supports pre-replication complex (pre-RC) assembly, while cyclin A-associated kinase activates DNA synthesis. We show that cyclin E, but not A, is mounted upon the nuclear matrix in sub-nuclear foci in differentiated vertebrate cells, but not in undifferentiated cells or cancer cells. In murine embryonic stem cells, Xenopus embryos and human urothelial cells, cyclin E is recruited to the nuclear matrix as cells differentiate and this can be manipulated in vitro. This suggests that pre-RC assembly becomes spatially restricted as template usage is defined. Furthermore, failure to become restricted may contribute to the plasticity of cancer cells. | | 21109536
|
Trop2 expression contributes to tumor pathogenesis by activating the ERK MAPK pathway.
Cubas, R; Zhang, S; Li, M; Chen, C; Yao, Q
Molecular cancer
9
253
2010
Show Abstract
Trop2 is a cell-surface glycoprotein overexpressed by a variety of epithelial carcinomas with reported low to restricted expression in normal tissues. Expression of Trop2 has been associated with increased tumor aggressiveness, metastasis and decreased patient survival, but the signaling mechanisms mediated by Trop2 are still unknown. Here, we studied the effects murine Trop2 (mTrop2) exerted on tumor cellular functions and some of the signaling mechanisms activated by this oncogene.mTrop2 expression significantly increased tumor cell proliferation at low serum concentration, migration, foci formation and anchorage-independent growth. These in vitro characteristics translated to increased tumor growth in both subcutaneous and orthotopic pancreatic cancer murine models and also led to increased liver metastasis. mTrop2 expression also increased the levels of phosphorylated ERK1/2 mediating cell cycle progression by increasing the levels of cyclin D1 and cyclin E as well as downregulating p27. The activation of ERK was also observed in human pancreatic ductal epithelial cells and colorectal adenocarcinoma cells overexpressing human Trop2.These findings demonstrate some of the pathogenic effects mediated by mTrop2 expression on cancer cells and the importance of targeting this cell surface glycoprotein. This study also provides the first indication of a molecular signaling pathway activated by Trop2 which has important implications for cancer cell growth and survival. Full Text Article | | 20858281
|
The retinoblastoma gene Rb and its family member p130 suppress lung adenocarcinoma induced by oncogenic K-Ras.
Ho, VM; Schaffer, BE; Karnezis, AN; Park, KS; Sage, J
Oncogene
28
1393-9
2009
Show Abstract
Mutations of the retinoblastoma tumor suppressor gene RB are frequently observed in human cancers, but rarely in non-small cell lung carcinomas (NSCLCs). Emerging evidence also suggests that the RB-related gene p130 is inactivated in a subset of human NSCLCs. To directly test the specific tumor suppressor roles of RB and p130 in NSCLC, we crossed Rb and p130 conditional mutant mice to mice carrying a conditional oncogenic K-Ras allele. In this model, controlled oncogenic K-Ras activation leads to the development of adenocarcinoma, a major subtype of NSCLC. We found that loss of p130 accelerated the death of mice, providing direct evidence in vivo that p130 is a tumor suppressor gene, albeit a weak one in this context. Loss of Rb increased the efficiency of lung cancer initiation and resulted in the development of high-grade adenocarcinomas and rapid death. Thus, despite the low frequency of RB mutations in human NSCLCs and reports that K-Ras activation and loss of RB function are rarely found in the same human tumors, loss of Rb clearly cooperates with activation of oncogenic K-Ras in lung adenocarcinoma development in mice. | Western Blotting | 19151761
|
The cell cycle and DNA mismatch repair.
Allen G Schroering,Michael A Edelbrock,Timothy J Richards,Kandace J Williams
Experimental cell research
313
2007
Show Abstract
The DNA mismatch repair (MMR) pathway contributes to the fidelity of DNA synthesis and recombination by correcting mispaired nucleotides and insertion/deletion loops (IDLs). We have investigated whether MMR protein expression, activity, and subcellular location are altered during discrete phases of the cell cycle in mammalian cells. Two distinct methods have been used to demonstrate that although physiological MMR protein expression, mismatch binding, and nick-directed MMR activity within the nucleus are at highest levels during S phase, MMR is active throughout the cell cycle. Despite equal MMR nuclear protein concentrations in S and G(2) phases, mismatch binding and repair activities within G(2) are significantly lower, indicating a post-translational decrease in MMR activity specific to G(2). We further demonstrate that typical co-localization of MutSalpha to late S phase replication foci can be disrupted by 2 microM N-methyl-N'-nitro-N-nitrosoguanidine (MNNG). This concentration of MNNG does not decrease ongoing DNA synthesis nor induce cell cycle arrest until the second cell cycle, with long-term colony survival decreased by only 24%. These results suggest that low level alkylation damage can selectively disrupt MMR proofreading activity during DNA synthesis and potentially increase mutation frequency within surviving cells. | | 17157834
|
Liver-specific loss of beta-catenin results in delayed hepatocyte proliferation after partial hepatectomy.
Shigeki Sekine, Pedro J A Gutiérrez, Billy Yu-Ang Lan, Sandy Feng, Matthias Hebrok
Hepatology (Baltimore, Md.)
45
361-8
2007
Show Abstract
Recent studies have suggested that beta-catenin is involved in the regulation of hepatocyte proliferation in multiple contexts, including organ development and tumorigenesis. We explored the role of beta-catenin during liver regeneration using T cell factor/lymphoid enhancer factor (TCF/LEF)-reporter mice (TOPGal mice) and liver-specific beta-catenin knockout mice. Liver-specific beta-catenin knockout mice showed a delayed onset of DNA synthesis after hepatectomy, whereas recovery of liver mass was not affected. Among putative beta-catenin target genes examined, the induction of Ccnd1 expression was reduced, whereas the expression of Myc and Egfr was unaffected. Furthermore, cyclin D1 protein levels were not induced, and the expression of cyclins A, E, and proliferating cell nuclear antigen was delayed. Intriguingly, the analysis of TOPGal mice showed that hepatocytes with active TCF/LEF transcription are confined to the pericentral zone and are not increased in number during regeneration, indicating an uncoupling between beta-catenin/TCF signaling activity and hepatocyte proliferation. Conclusion: Our results indicate that beta-catenin is critical for the proper regulation of hepatocyte proliferation during liver regeneration; however, the activity of beta-catenin/TCF signaling does not correlate with hepatocyte proliferation, suggesting that this regulation might be indirect/secondary. | | 17256747
|
The Rho GTPase effector ROCK regulates cyclin A, cyclin D1, and p27Kip1 levels by distinct mechanisms.
Croft, DR; Olson, MF
Molecular and cellular biology
26
4612-27
2006
Show Abstract
The members of the Rho GTPase family are well known for their regulation of actin cytoskeletal structures. In addition, they influence progression through the cell cycle. The RhoA and RhoC proteins regulate numerous effector proteins, with a central and vital signaling role mediated by the ROCK I and ROCK II serine/threonine kinases. The requirement for ROCK function in the proliferation of numerous cell types has been revealed by studies utilizing ROCK-selective inhibitors such as Y-27632. However, the mechanisms by which ROCK signaling promotes cell cycle progression have not been thoroughly characterized. Using a conditionally activated ROCK-estrogen receptor fusion protein, we found that ROCK activation is sufficient to stimulate G1/S cell cycle progression in NIH 3T3 mouse fibroblasts. Further analysis revealed that ROCK acts via independent pathways to alter the levels of cell cycle regulatory proteins: cyclin D1 and p21(Cip1) elevation via Ras and the mitogen-activated protein kinase pathway, increased cyclin A via LIM kinase 2, and reduction of p27(Kip1) protein levels. Therefore, the influence of ROCK on cell cycle regulatory proteins occurs by multiple independent mechanisms. | | 16738326
|
Rescue of cyclin D1 deficiency by knockin cyclin E.
Geng, Y, et al.
Cell, 97: 767-77 (1999)
1999
Show Abstract
D-type cyclins and cyclin E represent two very distinct classes of mammalian G1 cyclins. We have generated a mouse strain in which the coding sequences of the cyclin D1 gene (Ccnd1) have been deleted and replaced by those of human cyclin E (CCNE). In the tissues and cells of these mice, the expression pattern of human cyclin E faithfully reproduces that normally associated with mouse cyclin D1. The replacement of cyclin D1 with cyclin E rescues all phenotypic manifestations of cyclin D1 deficiency and restores normal development in cyclin D1-dependent tissues. Thus, cyclin E can functionally replace cyclin D1. Our analyses suggest that cyclin E is the major downstream target of cyclin D1. | | 10380928
|
Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2.
Prall, O W, et al.
J. Biol. Chem., 272: 10882-94 (1997)
1997
Show Abstract
Estrogens induce cell proliferation in target tissues by stimulating progression through G1 phase of the cell cycle, but the underlying molecular targets remain undefined. To determine the role of the cyclin/cyclin-dependent kinase (CDK)/retinoblastoma protein (pRB) pathway in this response we treated MCF-7 breast cancer cells with the pure estrogen antagonist ICI 182780 to inhibit estrogen-induced gene expression and induce G1 phase arrest. Subsequent treatment with 17beta-estradiol resulted in the synchronous entry of cells into S phase commencing at 12 h. The proportion of cells in S phase reached a maximum of 60% at 21-24 h. Cells subsequently completed mitosis and entered a second semisynchronous round of replication. Entry into S phase was preceded by increased activity of both Cdk4 and cyclin E-Cdk2 and hyperphosphorylation of pRB, all within the first 3-6 h of estradiol treatment. The increase in Cdk4 activity was accompanied by increases in cyclin D1 mRNA and protein, indicating that an initiating event in the activation of Cdk4 was increased cyclin D1 gene expression. In contrast, the levels of Cdk2 and the CDK inhibitors p21 (WAF1/CIP1/SDI1) and p27 (KIP1) in total cell lysates and in cyclin E immunoprecipitates were unaltered at these early time points. However, an inhibitory activity was present in antiestrogen-pretreated cell lysates toward recombinant cyclin E-Cdk2 and was relieved by estradiol treatment. This activity was attributable predominantly to p21. These apparently conflicting data were resolved by performing gel filtration chromatography, which revealed that only a minority of cyclin E-Cdk2 complexes were active following estradiol treatment. Active complexes eluted at a higher molecular weight than inactive complexes, were relatively deficient in both p21 and p27, and contained Cdk2 with increased threonine 160 phosphorylation, consistent with a mechanism of activation of cyclin E-Cdk2 involving both reduced CDK inhibitor association and CDK-activating kinase-mediated phosphorylation of Cdk2. These results provide an explanation for the early activation of both cyclin D1-Cdk4 and cyclin E-Cdk2 complexes that accompany G1-S phase progression in response to estradiol. | | 9099745
|