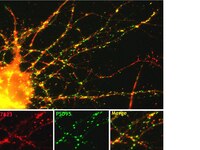
The cockayne syndrome B protein is essential for neuronal differentiation and neuritogenesis.
Ciaffardini, F; Nicolai, S; Caputo, M; Canu, G; Paccosi, E; Costantino, M; Frontini, M; Balajee, AS; Proietti-De-Santis, L
Cell death & disease
5
e1268
2014
Show Abstract
Cockayne syndrome (CS) is a progressive developmental and neurodegenerative disorder resulting in premature death at childhood and cells derived from CS patients display DNA repair and transcriptional defects. CS is caused by mutations in csa and csb genes, and patients with csb mutation are more prevalent. A hallmark feature of CSB patients is neurodegeneration but the precise molecular cause for this defect remains enigmatic. Further, it is not clear whether the neurodegenerative condition is due to loss of CSB-mediated functions in adult neurogenesis. In this study, we examined the role of CSB in neurogenesis by using the human neural progenitor cells that have self-renewal and differentiation capabilities. In this model system, stable CSB knockdown dramatically reduced the differentiation potential of human neural progenitor cells revealing a key role for CSB in neurogenesis. Neurite outgrowth, a characteristic feature of differentiated neurons, was also greatly abolished in CSB-suppressed cells. In corroboration with this, expression of MAP2 (microtubule-associated protein 2), a crucial player in neuritogenesis, was also impaired in CSB-suppressed cells. Consistent with reduced MAP2 expression in CSB-depleted neural cells, tandem affinity purification and chromatin immunoprecipitation studies revealed a potential role for CSB in the assembly of transcription complex on MAP2 promoter. Altogether, our data led us to conclude that CSB has a crucial role in coordinated regulation of transcription and chromatin remodeling activities that are required during neurogenesis. | Immunocytochemistry | 24874740
 |
Hippocampal dysregulation of synaptic plasticity-associated proteins with age-related cognitive decline.
Heather D VanGuilder,Julie A Farley,Han Yan,Colleen A Van Kirk,Matthew Mitschelen,William E Sonntag,Willard M Freeman
Neurobiology of disease
43
2011
Show Abstract
Age-related cognitive decline occurs without frank neurodegeneration and is the most common cause of memory impairment in aging individuals. With increasing longevity, cognitive deficits, especially in hippocampus-dependent memory processes, are increasing in prevalence. Nevertheless, the neurobiological basis of age-related cognitive decline remains unknown. While concerted efforts have led to the identification of neurobiological changes with aging, few age-related alterations have been definitively correlated to behavioral measures of cognitive decline. In this work, adult (12 months) and aged (28 months) rats were categorized by Morris water maze performance as Adult cognitively Intact, Aged cognitively Intact or Aged cognitively Impaired, and protein expression was examined in hippocampal synaptosome preparations. Previously described differences in synaptic expression of neurotransmission-associated proteins (Dnm1, Hpca, Stx1, Syn1, Syn2, Syp, SNAP25, VAMP2 and 14-3-3 eta, gamma, and zeta) were confirmed between Adult and Aged rats, with no further dysregulation associated with cognitive impairment. Proteins related to synaptic structural stability (MAP2, drebrin, Nogo-A) and activity-dependent signaling (PSD-95, 14-3-3θ, CaMKIIα) were up- and down-regulated, respectively, with cognitive impairment but were not altered with increasing age. Localization of MAP2, PSD-95, and CaMKIIα demonstrated protein expression alterations throughout the hippocampus. The altered expression of activity- and structural stability-associated proteins suggests that impaired synaptic plasticity is a distinct phenomenon that occurs with age-related cognitive decline, and demonstrates that cognitive decline is not simply an exacerbation of the aging phenotype. | | 21440628
|