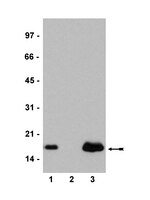
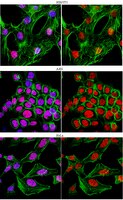
Genome-wide targeting of the epigenetic regulatory protein CTCF to gene promoters by the transcription factor TFII-I.
Peña-Hernández, R; Marques, M; Hilmi, K; Zhao, T; Saad, A; Alaoui-Jamali, MA; del Rincon, SV; Ashworth, T; Roy, AL; Emerson, BM; Witcher, M
Proceedings of the National Academy of Sciences of the United States of America
112
E677-86
2015
Show Abstract
CCCTC-binding factor (CTCF) is a key regulator of nuclear chromatin structure and gene regulation. The impact of CTCF on transcriptional output is highly varied, ranging from repression to transcriptional pausing and transactivation. The multifunctional nature of CTCF may be directed solely through remodeling chromatin architecture. However, another hypothesis is that the multifunctional nature of CTCF is mediated, in part, through differential association with protein partners having unique functions. Consistent with this hypothesis, our mass spectrometry analyses of CTCF interacting partners reveal a previously undefined association with the transcription factor general transcription factor II-I (TFII-I). Biochemical fractionation of CTCF indicates that a distinct CTCF complex incorporating TFII-I is assembled on DNA. Unexpectedly, we found that the interaction between CTCF and TFII-I is essential for directing CTCF to the promoter proximal regulatory regions of target genes across the genome, particularly at genes involved in metabolism. At genes coregulated by CTCF and TFII-I, we find knockdown of TFII-I results in diminished CTCF binding, lack of cyclin-dependent kinase 8 (CDK8) recruitment, and an attenuation of RNA polymerase II phosphorylation at serine 5. Phenotypically, knockdown of TFII-I alters the cellular response to metabolic stress. Our data indicate that TFII-I directs CTCF binding to target genes, and in turn the two proteins cooperate to recruit CDK8 and enhance transcription initiation. | | 25646466
 |
Dynamics of DOT1L localization and H3K79 methylation during meiotic prophase I in mouse spermatocytes.
Ontoso, D; Kauppi, L; Keeney, S; San-Segundo, PA
Chromosoma
123
147-64
2014
Show Abstract
During meiotic prophase I, interactions between maternal and paternal chromosomes, under checkpoint surveillance, establish connections between homologs that promote their accurate distribution to meiotic progeny. In human, faulty meiosis causes aneuploidy resulting in miscarriages and genetic diseases. Meiotic processes occur in the context of chromatin; therefore, histone post-translational modifications are expected to play important roles. Here, we report the cytological distribution of the evolutionarily conserved DOT1L methyltransferase and the different H3K79 methylation states resulting from its activity (mono-, di- and tri-methylation; H3K79me1, me2 and me3, respectively) during meiotic prophase I in mouse spermatocytes. In the wild type, whereas low amounts of H3K79me1 are rather uniformly present throughout prophase I, levels of DOT1L, H3K79me2 and H3K79me3 exhibit a notable increase from pachynema onwards, but with differential subnuclear distribution patterns. The heterochromatic centromeric regions and the sex body are enriched for H3K79me3. In contrast, H3K79me2 is present all over the chromatin, but is largely excluded from the sex body despite the accumulation of DOT1L. In meiosis-defective mouse mutants, the increase of DOT1L and H3K79me is blocked at the same stage where meiosis is arrested. H3K79me patterns, combined with the cytological analysis of the H3.3, γH2AX, macroH2A and H2A.Z histone variants, are consistent with a differential role for these epigenetic marks in male mouse meiotic prophase I. We propose that H3K79me2 is related to transcriptional reactivation on autosomes during pachynema, whereas H3K79me3 may contribute to the maintenance of repressive chromatin at centromeric regions and the sex body. | Immunofluorescence | 24105599
|
Phosphorylation and arginine methylation mark histone H2A prior to deposition during Xenopus laevis development.
Wang, WL; Anderson, LC; Nicklay, JJ; Chen, H; Gamble, MJ; Shabanowitz, J; Hunt, DF; Shechter, D
Epigenetics & chromatin
7
22
2014
Show Abstract
Stored, soluble histones in eggs are essential for early development, in particular during the maternally controlled early cell cycles in the absence of transcription. Histone post-translational modifications (PTMs) direct and regulate chromatin-templated transactions, so understanding the nature and function of pre-deposition maternal histones is essential to deciphering mechanisms of regulation of development, chromatin assembly, and transcription. Little is known about histone H2A pre-deposition modifications nor known about the transitions that occur upon the onset of zygotic control of the cell cycle and transcription at the mid-blastula transition (MBT).We isolated histones from staged Xenopus laevis oocytes, eggs, embryos, and assembled pronuclei to identify changes in histone H2A modifications prior to deposition and in chromatin. Soluble and chromatin-bound histones from eggs and embryos demonstrated distinct patterns of maternal and zygotic H2A PTMs, with significant pre-deposition quantities of S1ph and R3me1, and R3me2s. We observed the first functional distinction between H2A and H4 S1 phosphorylation, as we showed that H2A and H2A.X-F (also known as H2A.X.3) serine 1 (S1) is phosphorylated concomitant with germinal vesicle breakdown (GVBD) while H4 serine 1 phosphorylation occurs post-MBT. In egg extract H2A/H4 S1 phosphorylation is independent of the cell cycle, chromatin assembly, and DNA replication. H2AS1ph is highly enriched on blastula chromatin during repression of zygotic gene expression while H4S1ph is correlated with the beginning of maternal gene expression and the lengthening of the cell cycle, consistent with distinct biological roles for H2A and H4 S1 phosphorylation. We isolated soluble H2A and H2A.X-F from the egg and chromatin-bound in pronuclei and analyzed them by mass spectrometry analysis to quantitatively determine abundances of S1ph and R3 methylation. We show that H2A and H4 S1ph, R3me1 and R3me2s are enriched on nucleosomes containing both active and repressive histone PTMs in human A549 cells and Xenopus embryos.Significantly, we demonstrated that H2A phosphorylation and H4 arginine methylation form a new class of bona fide pre-deposition modifications in the vertebrate embryo. We show that S1ph and R3me containing chromatin domains are not correlated with H3 regulatory PTMs, suggesting a unique role for phosphorylation and arginine methylation. | Immunoblotting (Western) | 25302076
|
Histone H3.3 and its proteolytically processed form drive a cellular senescence programme.
Duarte, LF; Young, AR; Wang, Z; Wu, HA; Panda, T; Kou, Y; Kapoor, A; Hasson, D; Mills, NR; Ma'ayan, A; Narita, M; Bernstein, E
Nature communications
5
5210
2014
Show Abstract
The process of cellular senescence generates a repressive chromatin environment, however, the role of histone variants and histone proteolytic cleavage in senescence remains unclear. Here, using models of oncogene-induced and replicative senescence, we report novel histone H3 tail cleavage events mediated by the protease Cathepsin L. We find that cleaved forms of H3 are nucleosomal and the histone variant H3.3 is the preferred cleaved form of H3. Ectopic expression of H3.3 and its cleavage product (H3.3cs1), which lacks the first 21 amino acids of the H3 tail, is sufficient to induce senescence. Further, H3.3cs1 chromatin incorporation is mediated by the HUCA histone chaperone complex. Genome-wide transcriptional profiling revealed that H3.3cs1 facilitates transcriptional silencing of cell cycle regulators including RB/E2F target genes, likely via the permanent removal of H3K4me3. Collectively, our study identifies histone H3.3 and its proteolytically processed forms as key regulators of cellular senescence. | | 25394905
|
Mouse BAZ1A (ACF1) is dispensable for double-strand break repair but is essential for averting improper gene expression during spermatogenesis.
Dowdle, JA; Mehta, M; Kass, EM; Vuong, BQ; Inagaki, A; Egli, D; Jasin, M; Keeney, S
PLoS genetics
9
e1003945
2013
Show Abstract
ATP-dependent chromatin remodelers control DNA access for transcription, recombination, and other processes. Acf1 (also known as BAZ1A in mammals) is a defining subunit of the conserved ISWI-family chromatin remodelers ACF and CHRAC, first purified over 15 years ago from Drosophila melanogaster embryos. Much is known about biochemical properties of ACF and CHRAC, which move nucleosomes in vitro and in vivo to establish ordered chromatin arrays. Genetic studies in yeast, flies and cultured human cells clearly implicate these complexes in transcriptional repression via control of chromatin structures. RNAi experiments in transformed mammalian cells in culture also implicate ACF and CHRAC in DNA damage checkpoints and double-strand break repair. However, their essential in vivo roles in mammals are unknown. Here, we show that Baz1a-knockout mice are viable and able to repair developmentally programmed DNA double-strand breaks in the immune system and germ line, I-SceI endonuclease-induced breaks in primary fibroblasts via homologous recombination, and DNA damage from mitomycin C exposure in vivo. However, Baz1a deficiency causes male-specific sterility in accord with its high expression in male germ cells, where it displays dynamic, stage-specific patterns of chromosomal localization. Sterility is caused by pronounced defects in sperm development, most likely a consequence of massively perturbed gene expression in spermatocytes and round spermatids in the absence of BAZ1A: the normal spermiogenic transcription program is largely intact but more than 900 other genes are mis-regulated, primarily reflecting inappropriate up-regulation. We propose that large-scale changes in chromatin composition that occur during spermatogenesis create a window of vulnerability to promiscuous transcription changes, with an essential function of ACF and/or CHRAC chromatin remodeling activities being to safeguard against these alterations. | | 24244200
|
Synergism between DNA methylation and macroH2A1 occupancy in epigenetic silencing of the tumor suppressor gene p16(CDKN2A).
Barzily-Rokni, M; Friedman, N; Ron-Bigger, S; Isaac, S; Michlin, D; Eden, A
Nucleic acids research
39
1326-35
2011
Show Abstract
Promoter hypermethylation and heterochromatinization is a frequent event leading to gene inactivation and tumorigenesis. At the molecular level, inactivation of tumor suppressor genes in cancer has many similarities to the inactive X chromosome in female cells and is defined and maintained by DNA methylation and characteristic histone modifications. In addition, the inactive-X is marked by the histone macroH2A, a variant of H2A with a large non-histone region of unknown function. Studying tumor suppressor genes (TSGs) silenced in cancer cell lines, we find that when active, these promoters are associated with H2A.Z but become enriched for macroH2A1 once silenced. Knockdown of macroH2A1 was not sufficient for reactivation of silenced genes. However, when combined with DNA demethylation, macroH2A1 deficiency significantly enhanced reactivation of the tumor suppressor genes p16, MLH1 and Timp3 and inhibited cell proliferation. Our findings link macroH2A1 to heterochromatin of epigenetically silenced cancer genes and indicate synergism between macroH2A1 and DNA methylation in maintenance of the silenced state. | | 21030442
|
Molecular cloaking of H2A.Z on mortal DNA chromosomes during nonrandom segregation.
Yang Hoon Huh,James L Sherley
Stem cells (Dayton, Ohio)
29
2011
Show Abstract
Although nonrandom sister chromatid segregation is a singular property of distributed stem cells (DSCs) that are responsible for renewing and repairing mature vertebrate tissues, both its cellular function and its molecular mechanism remain unknown. This situation persists in part because of the lack of facile methods for detecting and quantifying nonrandom segregating cells and for identifying chromosomes with immortal DNA strands, the cellular molecules that signify nonrandom segregation. During nonrandom segregation, at each mitosis, asymmetrically self-renewing DSCs continuously cosegregate to themselves the set of chromosomes that contain immortal DNA strands, which are the oldest DNA strands. Here, we report the discovery of a molecular asymmetry between segregating sets of immortal chromosomes and opposed mortal chromosomes (i.e., containing the younger set of DNA template strands) that constitutes a new convenient biomarker for detection of cells undergoing nonrandom segregation and direct delineation of chromosomes that bear immortal DNA strands. In both cells engineered with DSC-specific properties and ex vivo-expanded mouse hair follicle stem cells, the histone H2A variant H2A.Z shows specific immunodetection on immortal DNA chromosomes. Cell fixation analyses indicate that H2A.Z is present on mortal chromosomes as well but is cloaked from immunodetection, and the cloaking entity is acid labile. The H2A.Z chromosomal asymmetry produced by molecular cloaking provides a first direct assay for nonrandom segregation and for chromosomes with immortal DNA strands. It also seems likely to manifest an important aspect of the underlying mechanism(s) responsible for nonrandom sister chromatid segregation in DSCs. | | 21905168
|
Histone crosstalk directed by H2B ubiquitination is required for chromatin boundary integrity.
Ma, MK; Heath, C; Hair, A; West, AG
PLoS genetics
7
e1002175
2011
Show Abstract
Genomic maps of chromatin modifications have provided evidence for the partitioning of genomes into domains of distinct chromatin states, which assist coordinated gene regulation. The maintenance of chromatin domain integrity can require the setting of boundaries. The HS4 insulator element marks the 3' boundary of a heterochromatin region located upstream of the chicken β-globin gene cluster. Here we show that HS4 recruits the E3 ligase RNF20/BRE1A to mediate H2B mono-ubiquitination (H2Bub1) at this insulator. Knockdown experiments show that RNF20 is required for H2Bub1 and processive H3K4 methylation. Depletion of RNF20 results in a collapse of the active histone modification signature at the HS4 chromatin boundary, where H2Bub1, H3K4 methylation, and hyperacetylation of H3, H4, and H2A.Z are rapidly lost. A remarkably similar set of events occurs at the HSA/HSB regulatory elements of the FOLR1 gene, which mark the 5' boundary of the same heterochromatin region. We find that persistent H2Bub1 at the HSA/HSB and HS4 elements is required for chromatin boundary integrity. The loss of boundary function leads to the sequential spreading of H3K9me2, H3K9me3, and H4K20me3 over the entire 50 kb FOLR1 and β-globin region and silencing of FOLR1 expression. These findings show that the HSA/HSB and HS4 boundary elements direct a cascade of active histone modifications that defend the FOLR1 and β-globin gene loci from the pervasive encroachment of an adjacent heterochromatin domain. We propose that many gene loci employ H2Bub1-dependent boundaries to prevent heterochromatin spreading. | | 21811414
|
Mapping and analysis of chromatin state dynamics in nine human cell types.
Ernst, J; Kheradpour, P; Mikkelsen, TS; Shoresh, N; Ward, LD; Epstein, CB; Zhang, X; Wang, L; Issner, R; Coyne, M; Ku, M; Durham, T; Kellis, M; Bernstein, BE
Nature
473
43-9
2011
Show Abstract
Chromatin profiling has emerged as a powerful means of genome annotation and detection of regulatory activity. The approach is especially well suited to the characterization of non-coding portions of the genome, which critically contribute to cellular phenotypes yet remain largely uncharted. Here we map nine chromatin marks across nine cell types to systematically characterize regulatory elements, their cell-type specificities and their functional interactions. Focusing on cell-type-specific patterns of promoters and enhancers, we define multicell activity profiles for chromatin state, gene expression, regulatory motif enrichment and regulator expression. We use correlations between these profiles to link enhancers to putative target genes, and predict the cell-type-specific activators and repressors that modulate them. The resulting annotations and regulatory predictions have implications for the interpretation of genome-wide association studies. Top-scoring disease single nucleotide polymorphisms are frequently positioned within enhancer elements specifically active in relevant cell types, and in some cases affect a motif instance for a predicted regulator, thus suggesting a mechanism for the association. Our study presents a general framework for deciphering cis-regulatory connections and their roles in disease. | | 21441907
|
Changes in the nuclear deposition of histone H2A variants during pre-implantation development in mice.
Nashun, B; Yukawa, M; Liu, H; Akiyama, T; Aoki, F
Development (Cambridge, England)
137
3785-94
2010
Show Abstract
Histone H2A has several variants, and changes in chromatin composition associated with their replacement might involve chromatin structure remodeling. We examined the dynamics of the canonical histone H2A and its three variants, H2A.X, H2A.Z and macroH2A, in the mouse during oogenesis and pre-implantation development when genome remodeling occurs. Immunocytochemistry with specific antibodies revealed that, although H2A and all variants were deposited in the nuclei of full-grown oocytes, only histone H2A.X was abundant in the pronuclei of one-cell embryos after fertilization, in contrast with the low abundance of histone H2A and the absence of H2A.Z. The decline in H2A and the depletion of H2A.Z and macroH2A after fertilization were confirmed using Flag epitope-tagged H2A, H2A.Z and macroH2A transgenic mouse lines. Microinjection experiments with mRNA encoding the Flag-tagged proteins revealed a similar pattern of nuclear incorporation of the H2A variants. Fusion protein experiments using H2A, H2A.Z and macroH2A fused with the C-terminal 23 amino acids of H2A.X showed that the C-terminal amino acids of H2A.X function specifically to target this variant histone into chromatin in embryos after fertilization and that the absence of H2A.Z and macroH2A from the chromatin is required for normal development. These results suggest that global changes in the composition of histone H2A variants in chromatin play a role in genome remodeling after fertilization. | Western Blotting | 20943707
|