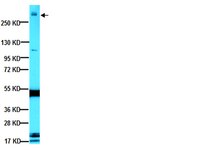
STIM2 protects hippocampal mushroom spines from amyloid synaptotoxicity.
Popugaeva, E; Pchitskaya, E; Speshilova, A; Alexandrov, S; Zhang, H; Vlasova, O; Bezprozvanny, I
Molecular neurodegeneration
10
37
2015
Show Abstract
Alzheimer disease (AD) is a disease of lost memories. Mushroom postsynaptic spines play a key role in memory storage, and loss of mushroom spines has been proposed to be linked to memory loss in AD. Generation of amyloidogenic peptides and accumulation of amyloid plaques is one of the pathological hallmarks of AD. It is important to evaluate effects of amyloid on stability of mushroom spines.In this study we used in vitro and in vivo models of amyloid synaptotoxicity to investigate effects of amyloid peptides on hippocampal mushroom spines. We discovered that application of Aβ42 oligomers to hippocampal cultures or injection of Aβ42 oligomers directly into hippocampal region resulted in reduction of mushroom spines and activity of synaptic calcium-calmodulin-dependent kinase II (CaMKII). We further discovered that expression of STIM2 protein rescued CaMKII activity and protected mushroom spines from amyloid toxicity in vitro and in vivo.Obtained results suggest that downregulation of STIM2-dependent stability of mushroom spines and reduction in activity of synaptic CaMKII is a mechanism of hippocampal synaptic loss in AD model of amyloid synaptotoxicity and that modulators/activators of this pathway may have a potential therapeutic value for treatment of AD. | | | 26275606
 |
HIV-1 Tat alters neuronal autophagy by modulating autophagosome fusion to the lysosome: implications for HIV-associated neurocognitive disorders.
Fields, J; Dumaop, W; Eleuteri, S; Elueteri, S; Campos, S; Serger, E; Trejo, M; Kosberg, K; Adame, A; Spencer, B; Rockenstein, E; He, JJ; Masliah, E
The Journal of neuroscience : the official journal of the Society for Neuroscience
35
1921-38
2015
Show Abstract
Antiretroviral therapy has increased the life span of HIV+ individuals; however, HIV-associated neurocognitive disorder (HAND) occurrence is increasing in aging HIV patients. Previous studies suggest HIV infection alters autophagy function in the aging CNS and HIV-1 proteins affect autophagy in monocyte-derived cells. Despite these findings, the mechanisms leading to dysregulated autophagy in the CNS remain unclear. Here we sought to determine how HIV Tat dysregulates autophagy in neurons. Tat caused a dose-dependent decrease in autophagosome markers, microtubule-associated protein-1 light chain β II (LC3II), and sequestosome 1(SQSTM1), in a membrane-enriched fraction, suggesting Tat increases autophagic degradation. Bafilomycin A1 increased autophagosome number, LC3II, and SQSTM1 accumulation; Tat cotreatment diminished this effect. Tat had no effect when 3-methyladenine or knockdown of beclin 1 blocked early stages of autophagy. Tat increased numbers of LC3 puncta and resulted in the formation of abnormal autophagosomes in vitro. Likewise, in vivo studies in GFAP-Tat tg mice showed increased autophagosome accumulation in neurons, altered LC3II levels, and neurodegeneration. These effects were reversed by rapamycin treatment. Tat colocalized with autophagosome and lysosomal markers and enhanced the colocalization of autophagosome with lysosome markers. Furthermore, co-IP studies showed that Tat interacts with lysosomal-associated membrane protein 2A (LAMP2A) in vitro and in vivo, and LAMP2A overexpression reduces Tat-induced neurotoxicity. Hence, Tat protein may induce autophagosome and lysosome fusion through interaction with LAMP2A leading to abnormal neuronal autophagy function and dysregulated degradation of critical intracellular components. Therapies targeting Tat-mediated autophagy alterations may decrease neurodegeneration in aging patients with HAND. | | | 25653352
|
MHC class I limits hippocampal synapse density by inhibiting neuronal insulin receptor signaling.
Dixon-Salazar, TJ; Fourgeaud, L; Tyler, CM; Poole, JR; Park, JJ; Boulanger, LM
The Journal of neuroscience : the official journal of the Society for Neuroscience
34
11844-56
2014
Show Abstract
Proteins of the major histocompatibility complex class I (MHCI) negatively regulate synapse density in the developing vertebrate brain (Glynn et al., 2011; Elmer et al., 2013; Lee et al., 2014), but the underlying mechanisms remain largely unknown. Here we identify a novel MHCI signaling pathway that involves the inhibition of a known synapse-promoting factor, the insulin receptor. Dominant-negative insulin receptor constructs decrease synapse density in the developing Xenopus visual system (Chiu et al., 2008), and insulin receptor activation increases dendritic spine density in mouse hippocampal neurons in vitro (Lee et al., 2011). We find that genetically reducing cell surface MHCI levels increases synapse density selectively in regions of the hippocampus where insulin receptors are expressed, and occludes the neuronal insulin response by de-repressing insulin receptor signaling. Pharmacologically inhibiting insulin receptor signaling in MHCI-deficient animals rescues synapse density, identifying insulin receptor signaling as a critical mediator of the tonic inhibitory effects of endogenous MHCI on synapse number. Insulin receptors co-immunoprecipitate MHCI from hippocampal lysates, and MHCI unmasks a cytoplasmic epitope of the insulin receptor that mediates downstream signaling. These results identify an important role for an MHCI-insulin receptor signaling pathway in circuit patterning in the developing brain, and suggest that changes in MHCI expression could unexpectedly regulate neuronal insulin sensitivity in the aging and diseased brain. | | | 25164678
|
Ischemia induces different levels of hypoxia inducible factor-1α protein expression in interneurons and pyramidal neurons.
Ramamoorthy, P; Shi, H
Acta neuropathologica communications
2
51
2014
Show Abstract
Pyramidal (glutamatergic) neurons and interneurons are morphologically and functionally well defined in the central nervous system. Although it is known that glutamatergic neurons undergo immediate cell death whereas interneurons are insensitive or survive longer during cerebral ischemia, the protection mechanisms responsible for this interneuronal survival are not well understood. Hypoxia inducible factor-1 (HIF-1) plays an important role in protecting neurons from hypoxic/ischemic insults. Here, we studied the expression of HIF-1α, the regulatable subunit of HIF-1, in the different neuronal phenotypes under in vitro and in vivo ischemia.In a primary cortical culture, HIF-1α expression was observed in neuronal somata after hypoxia (1% oxygen) in the presence of 5 or 25 mM glucose but not under normoxia (21% oxygen). Interestingly, only certain MAP2-positive neurons containing round somata (interneuron-like morphology) co-localized with HIF-1α staining. Other neurons such as pyramidal-like neurons showed no expression of HIF-1α under either normoxia or hypoxia. The HIF-1α positive neurons were GAD65/67 positive, confirming that they were interneuron-type cells. The HIF-1α expressing GAD65/67-positive neurons also possessed high levels of glutathione. We further demonstrated that ischemia induced significant HIF-1α expression in interneurons but not in pyramidal neurons in a rat model of middle cerebral artery occlusion.These results suggest that HIF-1α protein expression induced by ischemia is neuron-type specific and that this specificity may be related to the intracellular level of glutathione (GSH). | Immunofluorescence | | 24887017
|
SDF1 reduces interneuron leading process branching through dual regulation of actin and microtubules.
Lysko, DE; Putt, M; Golden, JA
The Journal of neuroscience : the official journal of the Society for Neuroscience
34
4941-62
2014
Show Abstract
Normal cerebral cortical function requires a highly ordered balance between projection neurons and interneurons. During development these two neuronal populations migrate from distinct progenitor zones to form the cerebral cortex, with interneurons originating in the more distant ganglionic eminences. Moreover, deficits in interneurons have been linked to a variety of neurodevelopmental disorders underscoring the importance of understanding interneuron development and function. We, and others, have identified SDF1 signaling as one important modulator of interneuron migration speed and leading process branching behavior in mice, although how SDF1 signaling impacts these behaviors remains unknown. We previously found SDF1 inhibited leading process branching while increasing the rate of migration. We have now mechanistically linked SDF1 modulation of leading process branching behavior to a dual regulation of both actin and microtubule organization. We find SDF1 consolidates actin at the leading process tip by de-repressing calpain protease and increasing proteolysis of branched-actin-supporting cortactin. Additionally, SDF1 stabilizes the microtubule array in the leading process through activation of the microtubule-associated protein doublecortin (DCX). DCX stabilizes the microtubule array by bundling microtubules within the leading process, reducing branching. These data provide mechanistic insight into the regulation of interneuron leading process dynamics during neuronal migration in mice and provides insight into how cortactin and DCX, a known human neuronal migration disorder gene, participate in this process. | | | 24695713
|
Neurotrophins regulate ApoER2 proteolysis through activation of the Trk signaling pathway.
Larios, JA; Jausoro, I; Benitez, ML; Bronfman, FC; Marzolo, MP
BMC neuroscience
15
108
2014
Show Abstract
ApoER2 and the neurotrophin receptors Trk and p75(NTR) are expressed in the CNS and regulate key functional aspects of neurons, including development, survival, and neuronal function. It is known that both ApoER2 and p75(NTR) are processed by metalloproteinases, followed by regulated intramembrane proteolysis. TrkA activation by nerve growth factor (NGF) increases the proteolytic processing of p75(NTR) mediated by ADAM17. Reelin induces the sheeding of ApoER2 ectodomain depending on metalloproteinase activity. However, it is not known if there is a common regulation mechanism for processing these receptors.We found that TrkA activation by NGF in PC12 cells induced ApoER2 processing, which was dependent on TrkA activation and metalloproteinases. NGF-induced ApoER2 proteolysis was independent of mitogen activated protein kinase activity and of phosphatidylinositol-3 kinase activity. In contrast, the basal proteolysis of ApoER2 increased when both kinases were pharmacologically inhibited. The ApoER2 ligand reelin regulated the proteolytic processing of its own receptor but not of p75(NTR). Finally, in primary cortical neurons, which express both ApoER2 and TrkB, we found that the proteolysis of ApoER2 was also regulated by brain-derived growth factor (BDNF).Our results highlight a novel relationship between neurotrophins and the reelin-ApoER2 system, suggesting that these two pathways might be linked to regulate brain development, neuronal survival, and some pathological conditions. | | | 25233900
|
Olfactory deficits in Niemann-Pick type C1 (NPC1) disease.
Hovakimyan, M; Meyer, A; Lukas, J; Luo, J; Gudziol, V; Hummel, T; Rolfs, A; Wree, A; Witt, M
PloS one
8
e82216
2013
Show Abstract
Niemann-Pick type C disease (NPC) is a rare autosomal recessive lipid storage disease characterized by progressive neurodegeneration. As only a few studies have been conducted on the impact of NPC on sensory systems, we used a mutant mouse model (NPC1(-/-)) to examine the effects of this disorder to morphologically distinct regions of the olfactory system, namely the olfactory epithelium (OE) and olfactory bulb (OB).For structural and functional analysis immunohistochemistry, electron microscopy, western blotting, and electrophysiology have been applied. For histochemistry and western blotting, we used antibodies against a series of neuronal and glia marker proteins, as well as macrophage markers. NPC1(-/-) animals present myelin-like lysosomal deposits in virtually all types of cells of the peripheral and central olfactory system. Especially supporting cells of the OE and central glia cells are affected, resulting in pronounced astrocytosis and microgliosis in the OB and other olfactory cortices. Up-regulation of Galectin-3, Cathepsin D and GFAP in the cortical layers of the OB underlines the critical role and location of the OB as a possible entrance gate for noxious substances. Unmyelinated olfactory afferents of the lamina propria seem less affected than ensheathing cells. Supporting the structural findings, electro-olfactometry of the olfactory mucosa suggests that NPC1(-/-) animals exhibit olfactory and trigeminal deficits.Our data demonstrate a pronounced neurodegeneration and glia activation in the olfactory system of NPC1(-/-), which is accompanied by sensory deficits. | | | 24391715
|
Automated imaging system for fast quantitation of neurons, cell morphology and neurite morphometry in vivo and in vitro.
Tapias, V; Greenamyre, JT; Watkins, SC
Neurobiology of disease
54
158-68
2013
Show Abstract
Quantitation of neurons using stereologic approaches reduces bias and systematic error, but is time-consuming and labor-intensive. Accurate methods for quantifying neurons in vitro are lacking; conventional methodologies are limited in reliability and application. The morphological properties of the soma and neurites are a key aspect of neuronal phenotype and function, but the assays commonly used in such evaluations are beset with several methodological drawbacks. Herein we describe automated techniques to quantify the number and morphology of neurons (or any cell type, e.g., astrocytes) and their processes with high speed and accuracy. Neuronal quantification from brain tissue using a motorized stage system yielded results that were statistically comparable to those generated by stereology. The approach was then adapted for in vitro neuron and neurite outgrowth quantification. To determine the utility of our methods, rotenone was used as a neurotoxicant leading to morphological changes in neurons and cell death, astrocytic activation, and loss of neurites. Importantly, our technique counted about 8 times as many neurons in less than 5-10% of the time taken by manual stereological analysis. | Immunohistochemistry | | 23220621
|
Sorafenib selectively depletes human glioblastoma tumor-initiating cells from primary cultures.
Carra, E; Barbieri, F; Marubbi, D; Pattarozzi, A; Favoni, RE; Florio, T; Daga, A
Cell cycle (Georgetown, Tex.)
12
491-500
2013
Show Abstract
Glioblastomas are grade IV brain tumors characterized by high aggressiveness and invasiveness, giving patients a poor prognosis. We investigated the effects of the multi-kinase inhibitor sorafenib on six cultures isolated from human glioblastomas and maintained in tumor initiating cells-enriching conditions. These cell subpopulations are thought to be responsible for tumor recurrence and radio- and chemo-resistance, representing the perfect target for glioblastoma therapy. Sorafenib reduces proliferation of glioblastoma cultures, and this effect depends, at least in part, on the inhibition of PI3K/Akt and MAPK pathways, both involved in gliomagenesis. Sorafenib significantly induces apoptosis/cell death via downregulation of the survival factor Mcl-1. We provide evidence that sorafenib has a selective action on glioblastoma stem cells, causing enrichment of cultures in differentiated cells, downregulation of the expression of stemness markers required to maintain malignancy (nestin, Olig2 and Sox2) and reducing cell clonogenic ability in vitro and tumorigenic potential in vivo. The selectivity of sorafenib effects on glioblastoma stem cells is confirmed by the lower sensitivity of glioblastoma cultures after differentiation as compared with the undifferentiated counterpart. Since current GBM therapy enriches the tumor in cancer stem cells, the evidence of a selective action of sorafenib on these cells is therapeutically relevant, even if, so far, results from first phase II clinical trials did not demonstrate its efficacy. | | | 23324350
|
Regulation of Wnt signaling by nociceptive input in animal models.
Shi, Y; Yuan, S; Li, B; Wang, J; Carlton, SM; Chung, K; Chung, JM; Tang, SJ
Molecular pain
8
47
2012
Show Abstract
Central sensitization-associated synaptic plasticity in the spinal cord dorsal horn (SCDH) critically contributes to the development of chronic pain, but understanding of the underlying molecular pathways is still incomplete. Emerging evidence suggests that Wnt signaling plays a crucial role in regulation of synaptic plasticity. Little is known about the potential function of the Wnt signaling cascades in chronic pain development.Fluorescent immunostaining results indicate that β-catenin, an essential protein in the canonical Wnt signaling pathway, is expressed in the superficial layers of the mouse SCDH with enrichment at synapses in lamina II. In addition, Wnt3a, a prototypic Wnt ligand that activates the canonical pathway, is also enriched in the superficial layers. Immunoblotting analysis indicates that both Wnt3a a β-catenin are up-regulated in the SCDH of various mouse pain models created by hind-paw injection of capsaicin, intrathecal (i.t.) injection of HIV-gp120 protein or spinal nerve ligation (SNL). Furthermore, Wnt5a, a prototypic Wnt ligand for non-canonical pathways, and its receptor Ror2 are also up-regulated in the SCDH of these models.Our results suggest that Wnt signaling pathways are regulated by nociceptive input. The activation of Wnt signaling may regulate the expression of spinal central sensitization during the development of acute and chronic pain. | Immunofluorescence | | 22713358
|