Establishment of Trophectoderm Cell Lines from Buffalo (Bubalus bubalis) Embryos of Different Sources and Examination of In Vitro Developmental Competence, Quality, Epigenetic Status and Gene Expression in Cloned Embryos Derived from Them.
Mohapatra, SK; Sandhu, A; Singh, KP; Singla, SK; Chauhan, MS; Manik, R; Palta, P
PloS one
10
e0129235
2015
Show Abstract
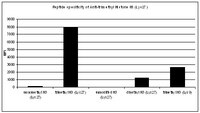
Despite being successfully used to produce live offspring in many species, somatic cell nuclear transfer (NT) has had a limited applicability due to very low (greater than 1%) live birth rate because of a high incidence of pregnancy failure, which is mainly due to placental dysfunction. Since this may be due to abnormalities in the trophectoderm (TE) cell lineage, TE cells can be a model to understand the placental growth disorders seen after NT. We isolated and characterized buffalo TE cells from blastocysts produced by in vitro fertilization (TE-IVF) and Hand-made cloning (TE-HMC), and compared their growth characteristics and gene expression, and developed a feeder-free culture system for their long-term culture. The TE-IVF cells were then used as donor cells to produce HMC embryos following which their developmental competence, quality, epigenetic status and gene expression were compared with those of HMC embryos produced using fetal or adult fibroblasts as donor cells. We found that although TE-HMC and TE-IVF cells have a similar capability to grow in culture, significant differences exist in gene expression levels between them and between IVF and HMC embryos from which they are derived, which may have a role in the placental abnormalities associated with NT pregnancies. Although TE cells can be used as donor cells for producing HMC blastocysts, their developmental competence and quality is lower than that of blastocysts produced from fetal or adult fibroblasts. The epigenetic status and expression level of many important genes is different in HMC blastocysts produced using TE cells or fetal or adult fibroblasts or those produced by IVF. | 26053554
 |
MAF1 represses CDKN1A through a Pol III-dependent mechanism.
Lee, YL; Li, YC; Su, CH; Chiao, CH; Lin, IH; Hsu, MT
eLife
4
e06283
2015
Show Abstract
MAF1 represses Pol III-mediated transcription by interfering with TFIIIB and Pol III. Herein, we found that MAF1 knockdown induced CDKN1A transcription and chromatin looping concurrently with Pol III recruitment. Simultaneous knockdown of MAF1 with Pol III or BRF1 (subunit of TFIIIB) diminished the activation and looping effect, which indicates that recruiting Pol III was required for activation of Pol II-mediated transcription and chromatin looping. Chromatin-immunoprecipitation analysis after MAF1 knockdown indicated enhanced binding of Pol III and BRF1, as well as of CFP1, p300, and PCAF, which are factors that mediate active histone marks, along with the binding of TATA binding protein (TBP) and POLR2E to the CDKN1A promoter. Simultaneous knockdown with Pol III abolished these regulatory events. Similar results were obtained for GDF15. Our results reveal a novel mechanism by which MAF1 and Pol III regulate the activity of a protein-coding gene transcribed by Pol II. | 26067234
|
Chromatin Signature Identifies Monoallelic Gene Expression Across Mammalian Cell Types.
Nag, A; Vigneau, S; Savova, V; Zwemer, LM; Gimelbrant, AA
G3 (Bethesda, Md.)
5
1713-20
2015
Show Abstract
Monoallelic expression of autosomal genes (MAE) is a widespread epigenetic phenomenon which is poorly understood, due in part to current limitations of genome-wide approaches for assessing it. Recently, we reported that a specific histone modification signature is strongly associated with MAE and demonstrated that it can serve as a proxy of MAE in human lymphoblastoid cells. Here, we use murine cells to establish that this chromatin signature is conserved between mouse and human and is associated with MAE in multiple cell types. Our analyses reveal extensive conservation in the identity of MAE genes between the two species. By analyzing MAE chromatin signature in a large number of cell and tissue types, we show that it remains consistent during terminal cell differentiation and is predominant among cell-type specific genes, suggesting a link between MAE and specification of cell identity. | 26092837
|
Hierarchical clustering of breast cancer methylomes revealed differentially methylated and expressed breast cancer genes.
Lin, IH; Chen, DT; Chang, YF; Lee, YL; Su, CH; Cheng, C; Tsai, YC; Ng, SC; Chen, HT; Lee, MC; Chen, HW; Suen, SH; Chen, YC; Liu, TT; Chang, CH; Hsu, MT
PloS one
10
e0118453
2015
Show Abstract
Oncogenic transformation of normal cells often involves epigenetic alterations, including histone modification and DNA methylation. We conducted whole-genome bisulfite sequencing to determine the DNA methylomes of normal breast, fibroadenoma, invasive ductal carcinomas and MCF7. The emergence, disappearance, expansion and contraction of kilobase-sized hypomethylated regions (HMRs) and the hypomethylation of the megabase-sized partially methylated domains (PMDs) are the major forms of methylation changes observed in breast tumor samples. Hierarchical clustering of HMR revealed tumor-specific hypermethylated clusters and differential methylated enhancers specific to normal or breast cancer cell lines. Joint analysis of gene expression and DNA methylation data of normal breast and breast cancer cells identified differentially methylated and expressed genes associated with breast and/or ovarian cancers in cancer-specific HMR clusters. Furthermore, aberrant patterns of X-chromosome inactivation (XCI) was found in breast cancer cell lines as well as breast tumor samples in the TCGA BRCA (breast invasive carcinoma) dataset. They were characterized with differentially hypermethylated XIST promoter, reduced expression of XIST, and over-expression of hypomethylated X-linked genes. High expressions of these genes were significantly associated with lower survival rates in breast cancer patients. Comprehensive analysis of the normal and breast tumor methylomes suggests selective targeting of DNA methylation changes during breast cancer progression. The weak causal relationship between DNA methylation and gene expression observed in this study is evident of more complex role of DNA methylation in the regulation of gene expression in human epigenetics that deserves further investigation. | 25706888
|
Genome-wide co-occupancy of AML1-ETO and N-CoR defines the t(8;21) AML signature in leukemic cells.
Trombly, DJ; Whitfield, TW; Padmanabhan, S; Gordon, JA; Lian, JB; van Wijnen, AJ; Zaidi, SK; Stein, JL; Stein, GS
BMC genomics
16
309
2015
Show Abstract
Many leukemias result from chromosomal rearrangements. The t(8;21) chromosomal translocation produces AML1-ETO, an oncogenic fusion protein that compromises the function of AML1, a transcription factor critical for myeloid cell differentiation. Because of the pressing need for new therapies in the treatment of acute myleoid leukemia, we investigated the genome-wide occupancy of AML1-ETO in leukemic cells to discover novel regulatory mechanisms involving AML-ETO bound genes.We report the co-localization of AML1-ETO with the N-CoR co-repressor to be primarily on genomic regions distal to transcriptional start sites (TSSs). These regions exhibit over-representation of the motif for PU.1, a key hematopoietic regulator and member of the ETS family of transcription factors. A significant discovery of our study is that genes co-occupied by AML1-ETO and N-CoR (e.g., TYROBP and LAPTM5) are associated with the leukemic phenotype, as determined by analyses of gene ontology and by the observation that these genes are predominantly up-regulated upon AML1-ETO depletion. In contrast, the AML1-ETO/p300 gene network is less responsive to AML1-ETO depletion and less associated with the differentiation block characteristic of leukemic cells. Furthermore, a substantial fraction of AML1-ETO/p300 co-localization occurs near TSSs in promoter regions associated with transcriptionally active loci.Our findings establish a novel and dominant t(8;21) AML leukemia signature characterized by occupancy of AML1-ETO/N-CoR at promoter-distal genomic regions enriched in motifs for myeloid differentiation factors, thus providing mechanistic insight into the leukemic phenotype. | 25928846
|
A balance between activating and repressive histone modifications regulates cystic fibrosis transmembrane conductance regulator (CFTR) expression in vivo.
Bergougnoux, A; Rivals, I; Liquori, A; Raynal, C; Varilh, J; Magalhães, M; Perez, MJ; Bigi, N; Des Georges, M; Chiron, R; Squalli-Houssaini, AS; Claustres, M; De Sario, A
Epigenetics
9
1007-17
2014
Show Abstract
The genetic mechanisms that regulate CFTR, the gene responsible for cystic fibrosis, have been widely investigated in cultured cells. However, mechanisms responsible for tissue-specific and time-specific expression are not completely elucidated in vivo. Through the survey of public databases, we found that the promoter of CFTR was associated with bivalent chromatin in human embryonic stem (ES) cells. In this work, we analyzed fetal (at different stages of pregnancy) and adult tissues and showed that, in digestive and lung tissues, which expressed CFTR, H3K4me3 was maintained in the promoter. Histone acetylation was high in the promoter and in two intronic enhancers, especially in fetal tissues. In contrast, in blood cells, which did not express CFTR, the bivalent chromatin was resolved (the promoter was labeled by the silencing mark H3K27me3). Cis-regulatory sequences were associated with lowly acetylated histones. We also provide evidence that the tissue-specific expression of CFTR is not regulated by dynamic changes of DNA methylation in the promoter. Overall, this work shows that a balance between activating and repressive histone modifications in the promoter and intronic enhancers results in the fine regulation of CFTR expression during development, thereby ensuring tissue specificity. | 24782114
|
Differentiation-dependent requirement of Tsix long non-coding RNA in imprinted X-chromosome inactivation.
Maclary, E; Buttigieg, E; Hinten, M; Gayen, S; Harris, C; Sarkar, MK; Purushothaman, S; Kalantry, S
Nature communications
5
4209
2014
Show Abstract
Imprinted X-inactivation is a paradigm of mammalian transgenerational epigenetic regulation resulting in silencing of genes on the paternally inherited X-chromosome. The preprogrammed fate of the X-chromosomes is thought to be controlled in cis by the parent-of-origin-specific expression of two opposing long non-coding RNAs, Tsix and Xist, in mice. Exclusive expression of Tsix from the maternal-X has implicated it as the instrument through which the maternal germline prevents inactivation of the maternal-X in the offspring. Here, we show that Tsix is dispensable for inhibiting Xist and X-inactivation in the early embryo and in cultured stem cells of extra-embryonic lineages. Tsix is instead required to prevent Xist expression as trophectodermal progenitor cells differentiate. Despite induction of wild-type Xist RNA and accumulation of histone H3-K27me3, many Tsix-mutant X-chromosomes fail to undergo ectopic X-inactivation. We propose a novel model of lncRNA function in imprinted X-inactivation that may also apply to other genomically imprinted loci. | 24979243
|
Structural basis of a histone H3 lysine 4 demethylase required for stem elongation in rice.
Chen, Q; Chen, X; Wang, Q; Zhang, F; Lou, Z; Zhang, Q; Zhou, DX
PLoS genetics
9
e1003239
2013
Show Abstract
Histone lysine methylation is an important epigenetic modification in regulating chromatin structure and gene expression. Histone H3 lysine 4 methylation (H3K4me), which can be in a mono-, di-, or trimethylated state, has been shown to play an important role in gene expression involved in plant developmental control and stress adaptation. However, the resetting mechanism of this epigenetic modification is not yet fully understood. In this work, we identified a JmjC domain-containing protein, JMJ703, as a histone lysine demethylase that specifically reverses all three forms of H3K4me in rice. Loss-of-function mutation of the gene affected stem elongation and plant growth, which may be related to increased expression of cytokinin oxidase genes in the mutant. Analysis of crystal structure of the catalytic core domain (c-JMJ703) of the protein revealed a general structural similarity with mammalian and yeast JMJD2 proteins that are H3K9 and H3K36 demethylases. However, several specific features were observed in the structure of c-JMJ703. Key residues that interact with cofactors Fe(II) and N-oxalylglycine and the methylated H3K4 substrate peptide were identified and were shown to be essential for the demethylase activity in vivo. Several key residues are specifically conserved in known H3K4 demethylases, suggesting that they may be involved in the specificity for H3K4 demethylation. | 23357881
|
BRG1 promotes survival of UV-irradiated melanoma cells by cooperating with MITF to activate the melanoma inhibitor of apoptosis gene.
Saladi, SV; Wong, PG; Trivedi, AR; Marathe, HG; Keenen, B; Aras, S; Liew, ZQ; Setaluri, V; de la Serna, IL
Pigment cell & melanoma research
26
377-91
2013
Show Abstract
Microphthalmia-associated transcription factor (MITF) is a survival factor in melanocytes and melanoma cells. MITF regulates expression of antiapoptotic genes and promotes lineage-specific survival in response to ultraviolet (UV) radiation and to chemotherapeutics. SWI/SNF chromatin-remodeling enzymes interact with MITF to regulate MITF target gene expression. We determined that the catalytic subunit, BRG1, of the SWI/SNF complex protects melanoma cells against UV-induced death. BRG1 prevents apoptosis in UV-irradiated melanoma cells by activating expression of the melanoma inhibitor of apoptosis (ML-IAP). Down-regulation of ML-IAP compromises BRG1-mediated survival of melanoma cells in response to UV radiation. BRG1 regulates ML-IAP expression by cooperating with MITF to promote transcriptionally permissive chromatin structure on the ML-IAP promoter. The alternative catalytic subunit, BRM, and the BRG1-associated factor, BAF180, were found to be dispensable for elevated expression of ML-IAP in melanoma cells. Thus, we illuminate a lineage-specific mechanism by which a specific SWI/SNF subunit, BRG1, modulates the cellular response to DNA damage by regulating an antiapoptotic gene and implicate this subunit of the SWI/SNF complex in mediating the prosurvival function of MITF. | 23480510
|
Development of multiple cell-based assays for the detection of histone H3 Lys27 trimethylation (H3K27me3).
Qian, J; Lu, L; Wu, J; Ma, H
Assay and drug development technologies
11
449-56
2013
Show Abstract
Posttranslational modification of histone proteins in eukaryotes plays an important role in gene transcription and chromatin structure. Dysregulation of the enzymes involved in histone modification has been linked to many cancer forms, making this target class a potential new area for therapeutics. A reliable assay to monitor small-molecule inhibition of various epigenetic enzymes should play a critical role in drug discovery to fight cancer. However, it has been challenging to develop cell-based assays for high-throughput screening (HTS) and compound profiling. Recently, two homogeneous cell-based assay kits using the AlphaLISA(®) and LanthaScreen(®) technologies to detect trimethyl histone H3 Lysine 27 have become commercially available, and a heterogeneous cell assay with modified dissociation-enhanced lanthanide fluorescence immunoassay (DELFIA(®)) format has been reported. To compare their pros and cons, we evaluated, optimized, and validated these three assay formats in three different cell lines and compared their activities with traditional Western blot detection of histone methylation inhibition by using commercial and in-house small-molecule inhibitors. Our data indicate that, although all four formats produced acceptable results, the homogeneous AlphaLISA assay was best suited for HTS and compound profiling due to its wider window and ease of automation. The DELFIA and Western blot assays were useful as validation tools to confirm the cell activities and eliminate potential false-positive compounds. | 23992119
|