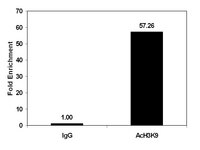
ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat.
Meidhof, S; Brabletz, S; Lehmann, W; Preca, BT; Mock, K; Ruh, M; Schüler, J; Berthold, M; Weber, A; Burk, U; Lübbert, M; Puhr, M; Culig, Z; Wellner, U; Keck, T; Bronsert, P; Küsters, S; Hopt, UT; Stemmler, MP; Brabletz, T
EMBO molecular medicine
7
831-47
2015
Show Abstract
Therapy resistance is a major clinical problem in cancer medicine and crucial for disease relapse and progression. Therefore, the clinical need to overcome it, particularly for aggressive tumors such as pancreatic cancer, is very high. Aberrant activation of an epithelial-mesenchymal transition (EMT) and an associated cancer stem cell phenotype are considered a major cause of therapy resistance. Particularly, the EMT-activator ZEB1 was shown to confer stemness and resistance. We applied a systematic, stepwise strategy to interfere with ZEB1 function, aiming to overcome drug resistance. This led to the identification of both its target gene miR-203 as a major drug sensitizer and subsequently the class I HDAC inhibitor mocetinostat as epigenetic drug to interfere with ZEB1 function, restore miR-203 expression, repress stemness properties, and induce sensitivity against chemotherapy. Thereby, mocetinostat turned out to be more effective than other HDAC inhibitors, such as SAHA, indicating the relevance of the screening strategy. Our data encourage the application of mechanism-based combinations of selected epigenetic drugs with standard chemotherapy for the rational treatment of aggressive solid tumors, such as pancreatic cancer. | | 25872941
 |
The histone acetyltransferase MOF activates hypothalamic polysialylation to prevent diet-induced obesity in mice.
Brenachot, X; Rigault, C; Nédélec, E; Laderrière, A; Khanam, T; Gouazé, A; Chaudy, S; Lemoine, A; Datiche, F; Gascuel, J; Pénicaud, L; Benani, A
Molecular metabolism
3
619-29
2014
Show Abstract
Overfeeding causes rapid synaptic remodeling in hypothalamus feeding circuits. Polysialylation of cell surface molecules is a key step in this neuronal rewiring and allows normalization of food intake. Here we examined the role of hypothalamic polysialylation in the long-term maintenance of body weight, and deciphered the molecular sequence underlying its nutritional regulation. We found that upon high fat diet (HFD), reduced hypothalamic polysialylation exacerbated the diet-induced obese phenotype in mice. Upon HFD, the histone acetyltransferase MOF was rapidly recruited on the St8sia4 polysialyltransferase-encoding gene. Mof silencing in the mediobasal hypothalamus of adult mice prevented activation of the St8sia4 gene transcription, reduced polysialylation, altered the acute homeostatic feeding response to HFD and increased the body weight gain. These findings indicate that impaired hypothalamic polysialylation contribute to the development of obesity, and establish a role for MOF in the brain control of energy balance. | | 25161885
|
Transgenerational phenotypic and epigenetic changes in response to heat stress in Arabidopsis thaliana.
Migicovsky, Z; Yao, Y; Kovalchuk, I
Plant signaling & behavior
9
e27971
2014
Show Abstract
Exposure to heat stress causes physiological and epigenetic changes in plants, which may also be altered in the progeny. We compared the progeny of stressed and control Arabidopsis thaliana wild type and Dicer-like mutant dcl2, dcl3, and dcl4 plants for variations in physiology and molecular profile, including global genome methylation, mRNA levels, and histone modifications in the subset of differentially expressed genes at normal conditions and in response to heat stress. We found that the immediate progeny of heat-stressed plants had fewer, but larger leaves, and tended to bolt earlier. Transposon expression was elevated in the progeny of heat-stressed plants, and heat stress in the same generation tended to decrease global genome methylation. Progeny of stressed plants had increased expression of HSFA2, and reduction in MSH2, ROS1, and several SUVH genes. Gene expression positively correlated with permissive histone marks and negatively correlated with repressive marks. Overall, the progeny of heat stressed plants varied in both their physiology and epigenome and dcl2 and dcl3 mutants were partially deficient for these changes. | | 24513700
|
Deletion of a conserved cis-element in the Ifng locus highlights the role of acute histone acetylation in modulating inducible gene transcription.
Balasubramani, A; Winstead, CJ; Turner, H; Janowski, KM; Harbour, SN; Shibata, Y; Crawford, GE; Hatton, RD; Weaver, CT
PLoS genetics
10
e1003969
2014
Show Abstract
Differentiation-dependent regulation of the Ifng cytokine gene locus in T helper (Th) cells has emerged as an excellent model for functional study of distal elements that control lineage-specific gene expression. We previously identified a cis-regulatory element located 22 kb upstream of the Ifng gene (Conserved Non-coding Sequence -22, or CNS-22) that is a site for recruitment of the transcription factors T-bet, Runx3, NF-κB and STAT4, which act to regulate transcription of the Ifng gene in Th1 cells. Here, we report the generation of mice with a conditional deletion of CNS-22 that has enabled us to define the epigenetic and functional consequences of its absence. Deletion of CNS-22 led to a defect in induction of Ifng by the cytokines IL-12 and IL-18, with a more modest effect on induction via T-cell receptor activation. To better understand how CNS-22 and other Ifng CNSs regulated Ifng transcription in response to these distinct stimuli, we examined activation-dependent changes in epigenetic modifications across the extended Ifng locus in CNS-22-deficient T cells. We demonstrate that in response to both cytokine and TCR driven activation signals, CNS-22 and other Ifng CNSs recruit increased activity of histone acetyl transferases (HATs) that transiently enhance levels of histones H3 and H4 acetylation across the extended Ifng locus. We also demonstrate that activation-responsive increases in histone acetylation levels are directly linked to the ability of Ifng CNSs to acutely enhance Pol II recruitment to the Ifng promoter. Finally, we show that impairment in IL-12+IL-18 dependent induction of Ifng stems from the importance of CNS-22 in coordinating locus-wide levels of histone acetylation in response to these cytokines. These findings identify a role for acute histone acetylation in the enhancer function of distal conserved cis-elements that regulate of Ifng gene expression. | | 24415943
|
Depletion of sirtuin 1 (SIRT1) leads to epigenetic modifications of telomerase (TERT) gene in hepatocellular carcinoma cells.
Zhang, B; Chen, J; Cheng, AS; Ko, BC
PloS one
9
e84931
2014
Show Abstract
Sirtuin 1 (SIRT1) is a nicotinamide adenine dinucleotide (NAD)-dependent deacetylase that is implicated in plethora of biological processes, including metabolism, aging, stress response, and tumorigenesis. Telomerase (TERT) is essential for telomere maintenance. Activation of TERT is considered a crucial step in tumorigenesis, and therefore it is a potential therapeutic target against cancer. We have recently found that SIRT1 expression is highly elevated in hepatocellular carcinoma, and the depletion of SIRT1 leads to substantial reduction in TERT mRNA and protein expression. However, the underlying molecular mechanism of SIRT1-dependent TERT expression remains uncharacterized. Here, we elucidated if SIRT1 regulates TERT expression via transcriptional, epigenetic and post-transcriptional mechanisms. We report that depletion of SIRT1 does not lead to significant change in transcriptional activity and CpG methylation patterns of the TERT promoter, nor does it affect mRNA stability or 3'-UTR regulation of TERT. Intriguingly, depletion of SIRT1 is associated with substantial induction of acetylated histone H3-K9 and reduction of trimethyl H3-K9 at the TERT gene, which are known to be associated with gene activation. Our data revealed that SIRT1 regulates histone acetylation and methylation at the TERT promoter. We postulated that SIRT1 may regulate TERT expression via long-range interaction, or via yet unidentified histone modifications. | | 24416313
|
CREB-binding protein (CBP) regulates β-adrenoceptor (β-AR)-mediated apoptosis.
Lee, YY; Moujalled, D; Doerflinger, M; Gangoda, L; Weston, R; Rahimi, A; de Alboran, I; Herold, M; Bouillet, P; Xu, Q; Gao, X; Du, XJ; Puthalakath, H
Cell death and differentiation
20
941-52
2013
Show Abstract
Catecholamines regulate the β-adrenoceptor/cyclic AMP-regulated protein kinase A (cAMP/PKA) pathway. Deregulation of this pathway can cause apoptotic cell death and is implicated in a range of human diseases, such as neuronal loss during aging, cardiomyopathy and septic shock. The molecular mechanism of this process is, however, only poorly understood. Here we demonstrate that the β-adrenoceptor/cAMP/PKA pathway triggers apoptosis through the transcriptional induction of the pro-apoptotic BH3-only Bcl-2 family member Bim in tissues such as the thymus and the heart. In these cell types, the catecholamine-mediated apoptosis is abrogated by loss of Bim. Induction of Bim is driven by the transcriptional co-activator CBP (CREB-binding protein) together with the proto-oncogene c-Myc. Association of CBP with c-Myc leads to altered histone acetylation and methylation pattern at the Bim promoter site. Our findings have implications for understanding pathophysiology associated with a deregulated neuroendocrine system and for developing novel therapeutic strategies for these diseases. | Western Blotting | 23579242
|
Valproic acid inhibits the proliferation of cancer cells by re-expressing cyclin D2.
Witt, D; Burfeind, P; von Hardenberg, S; Opitz, L; Salinas-Riester, G; Bremmer, F; Schweyer, S; Thelen, P; Neesen, J; Kaulfuss, S
Carcinogenesis
34
1115-24
2013
Show Abstract
In this study, primary murine prostate cancer (PCa) cells were derived using the well-established TRAMP model. These PCa cells were treated with the histone deacetylase inhibitor, valproic acid (VPA), and we demonstrated that VPA treatment has an antimigrative, antiinvasive and antiproliferative effect on PCa cells. Using microarray analyses, we discovered several candidate genes that could contribute to the cellular effects we observed. In this study, we could demonstrate that VPA treatment of PCa cells causes the re-expression of cyclin D2, a known regulator that is frequently lost in PCa as we could show using immunohistochemical analyses on PCa specimens. We demonstrate that VPA specifically induces the re-expression of cyclin D2, one of the highly conserved D-type cyclin family members, in several cancer cell lines with weak or no cyclin D2 expression. Interestingly, VPA treatment had no effect in fibroblasts, which typically have high basal levels of cyclin D2 expression. The re-expression of cyclin D2 observed in PCa cells is activated by increased histone acetylation in the promoter region of the Ccnd2 gene and represents one underlying molecular mechanism of VPA treatment that inhibits the proliferation of cancer cells. Altogether, our results confirm that VPA is an anticancer therapeutic drug for the treatment of tumors with epigenetically repressed cyclin D2 expression. | | 23349020
|
Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts.
Huang, SK; Scruggs, AM; Donaghy, J; Horowitz, JC; Zaslona, Z; Przybranowski, S; White, ES; Peters-Golden, M
Cell death & disease
4
e621
2013
Show Abstract
Although the recruitment of fibroblasts to areas of injury is critical for wound healing, their subsequent apoptosis is necessary in order to prevent excessive scarring. Fibroproliferative diseases, such as pulmonary fibrosis, are often characterized by fibroblast resistance to apoptosis, but the mechanism(s) for this resistance remains elusive. Here, we employed a murine model of pulmonary fibrosis and cells from patients with idiopathic pulmonary fibrosis (IPF) to explore epigenetic mechanisms that may be responsible for the decreased expression of Fas, a cell surface death receptor whose expression has been observed to be decreased in pulmonary fibrosis. Murine pulmonary fibrosis was elicited by intratracheal injection of bleomycin. Fibroblasts cultured from bleomycin-treated mice exhibited decreased Fas expression and resistance to Fas-mediated apoptosis compared with cells from saline-treated control mice. Although there were no differences in DNA methylation, the Fas promoter in fibroblasts from bleomycin-treated mice exhibited decreased histone acetylation and increased histone 3 lysine 9 trimethylation (H3K9Me3). This was associated with increased histone deacetylase (HDAC)-2 and HDAC4 expression. Treatment with HDAC inhibitors increased Fas expression and restored susceptibility to Fas-mediated apoptosis. Fibroblasts from patients with IPF likewise exhibited decreased histone acetylation and increased H3K9Me3 at the Fas promoter and increased their expression of Fas in the presence of an HDAC inhibitor. These findings demonstrate the critical role of histone modifications in the development of fibroblast resistance to apoptosis in both a murine model and in patients with pulmonary fibrosis and suggest novel approaches to therapy for progressive fibroproliferative disorders. | Western Blotting | 23640463
|
Rev-erb-α modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy.
Woldt, E; Sebti, Y; Solt, LA; Duhem, C; Lancel, S; Eeckhoute, J; Hesselink, MK; Paquet, C; Delhaye, S; Shin, Y; Kamenecka, TM; Schaart, G; Lefebvre, P; Nevière, R; Burris, TP; Schrauwen, P; Staels, B; Duez, H
Nature medicine
19
1039-46
2013
Show Abstract
The nuclear receptor Rev-erb-α modulates hepatic lipid and glucose metabolism, adipogenesis and the inflammatory response in macrophages. We show here that Rev-erb-α is highly expressed in oxidative skeletal muscle and that its deficiency in muscle leads to reduced mitochondrial content and oxidative function, as well as upregulation of autophagy. These cellular effects resulted in both impaired mitochondrial biogenesis and increased clearance of this organelle, leading to compromised exercise capacity. On a molecular level, Rev-erb-α deficiency resulted in deactivation of the Lkb1-Ampk-Sirt1-Ppargc-1α signaling pathway. These effects were recapitulated in isolated fibers and in muscle cells after knockdown of the gene encoding Rev-erb-α, Nr1d1. In complementary experiments, Rev-erb-α overexpression in vitro increased the number of mitochondria and improved respiratory capacity, whereas muscle overexpression or pharmacological activation of Rev-erb-α in vivo increased exercise capacity. This study identifies Rev-erb-α as a pharmacological target that improves muscle oxidative function by modulating gene networks controlling mitochondrial number and function. | Western Blotting | 23852339
|
Epigenetic regulation of MDR1 gene through post-translational histone modifications in prostate cancer.
Henrique, R; Oliveira, AI; Costa, VL; Baptista, T; Martins, AT; Morais, A; Oliveira, J; Jerónimo, C
BMC Genomics
14
898
2013
Show Abstract
Multidrug resistance 1 (MDR1) gene encodes for an ATP binding cassette transporter--P-glycoprotein (P-gp)-- involved in chemoresistance to taxanes. MDR1 promoter methylation is frequent in prostate carcinoma (PCa), suggesting an epigenetic regulation but no functional correlation has been established. We aimed to elucidate the epigenetic mechanisms involved in MDR1 deregulation in PCa.MDR1 promoter methylation and P-gp expression were assessed in 121 PCa, 39 high-grade prostatic intraepithelial neoplasia (HGPIN), 28 benign prostatic hyperplasia (BPH) and 10 morphologically normal prostate tissue (NPT) samples, using quantitative methylation specific PCR and immunohistochemistry, respectively. PCa cell lines were exposed to a DNA methyltransferases inhibitor 5-aza-2'deoxycytidine (DAC) and histone deacetylases inhibitor trichostatin A (TSA). Methylation and histone posttranscriptional modifications status were characterized and correlated with mRNA and protein expression. MDR1 promoter methylation levels and frequency significantly increased from NPTs, to HGPIN and to PCa. Conversely, decreased or absent P-gp immunoexpression was observed in HGPIN and PCa, inversely correlating with methylation levels. Exposure to DAC alone did not alter significantly methylation levels, although increased expression was apparent. However, P-gp mRNA and protein re-expression were higher in cell lines exposed to TSA alone or combined with DAC. Accordingly, histone active marks H3Ac, H3K4me2, H3K4me3, H3K9Ac, and H4Ac were increased at the MDR1 promoter after exposure to TSA alone or combined with DAC.Our data suggests that, in prostate carcinogenesis, MDR1 downregulation is mainly due to histone post-translational modifications. This occurs concomitantly with aberrant promoter methylation, substantiating the association with P-gp decreased expression. | | 24344919
|