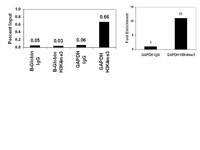
The gene signature in CCAAT-enhancer-binding protein α dysfunctional acute myeloid leukemia predicts responsiveness to histone deacetylase inhibitors.
Liss, A; Ooi, CH; Zjablovskaja, P; Benoukraf, T; Radomska, HS; Ju, C; Wu, M; Balastik, M; Delwel, R; Brdicka, T; Tan, P; Tenen, DG; Alberich-Jorda, M
Haematologica
99
697-705
2014
Show Abstract
C/EPBα proteins, encoded by the CCAAT-enhancer-binding protein α gene, play a crucial role in granulocytic development, and defects in this transcription factor have been reported in acute myeloid leukemia. Here, we defined the C/EBPα signature characterized by a set of genes up-regulated upon C/EBPα activation. We analyzed expression of the C/EBPα signature in a cohort of 525 patients with acute myeloid leukemia and identified a subset characterized by low expression of this signature. We referred to this group of patients as the C/EBPα dysfunctional subset. Remarkably, a large percentage of samples harboring C/EBPα biallelic mutations clustered within this subset. We hypothesize that re-activation of the C/EBPα signature in the C/EBPα dysfunctional subset could have therapeutic potential. In search for small molecules able to reverse the low expression of the C/EBPα signature we applied the connectivity map. This analysis predicted positive connectivity between the C/EBPα activation signature and histone deacetylase inhibitors. We showed that these inhibitors reactivate expression of the C/EBPα signature and promote granulocytic differentiation of primary samples from the C/EBPα dysfunctional subset harboring biallelic C/EBPα mutations. Altogether, our study identifies histone deacetylase inhibitors as potential candidates for the treatment of certain leukemias characterized by down-regulation of the C/EBPα signature. | 24162792
 |
Deletion of a conserved cis-element in the Ifng locus highlights the role of acute histone acetylation in modulating inducible gene transcription.
Balasubramani, A; Winstead, CJ; Turner, H; Janowski, KM; Harbour, SN; Shibata, Y; Crawford, GE; Hatton, RD; Weaver, CT
PLoS genetics
10
e1003969
2014
Show Abstract
Differentiation-dependent regulation of the Ifng cytokine gene locus in T helper (Th) cells has emerged as an excellent model for functional study of distal elements that control lineage-specific gene expression. We previously identified a cis-regulatory element located 22 kb upstream of the Ifng gene (Conserved Non-coding Sequence -22, or CNS-22) that is a site for recruitment of the transcription factors T-bet, Runx3, NF-κB and STAT4, which act to regulate transcription of the Ifng gene in Th1 cells. Here, we report the generation of mice with a conditional deletion of CNS-22 that has enabled us to define the epigenetic and functional consequences of its absence. Deletion of CNS-22 led to a defect in induction of Ifng by the cytokines IL-12 and IL-18, with a more modest effect on induction via T-cell receptor activation. To better understand how CNS-22 and other Ifng CNSs regulated Ifng transcription in response to these distinct stimuli, we examined activation-dependent changes in epigenetic modifications across the extended Ifng locus in CNS-22-deficient T cells. We demonstrate that in response to both cytokine and TCR driven activation signals, CNS-22 and other Ifng CNSs recruit increased activity of histone acetyl transferases (HATs) that transiently enhance levels of histones H3 and H4 acetylation across the extended Ifng locus. We also demonstrate that activation-responsive increases in histone acetylation levels are directly linked to the ability of Ifng CNSs to acutely enhance Pol II recruitment to the Ifng promoter. Finally, we show that impairment in IL-12+IL-18 dependent induction of Ifng stems from the importance of CNS-22 in coordinating locus-wide levels of histone acetylation in response to these cytokines. These findings identify a role for acute histone acetylation in the enhancer function of distal conserved cis-elements that regulate of Ifng gene expression. | 24415943
|
Alterations of epigenetic signatures in hepatocyte nuclear factor 4α deficient mouse liver determined by improved ChIP-qPCR and (h)MeDIP-qPCR assays.
Zhang, Q; Lei, X; Lu, H
PloS one
9
e84925
2014
Show Abstract
Hepatocyte nuclear factor 4α (HNF4α) is a liver-enriched transcription factor essential for liver development and function. In hepatocytes, HNF4α regulates a large number of genes important for nutrient/xenobiotic metabolism and cell differentiation and proliferation. Currently, little is known about the epigenetic mechanism of gene regulation by HNF4α. In this study, the global and specific alterations at the selected gene loci of representative histone modifications and DNA methylations were investigated in Hnf4a-deficient female mouse livers using the improved MeDIP-, hMeDIP- and ChIP-qPCR assay. Hnf4a deficiency significantly increased hepatic total IPed DNA fragments for histone H3 lysine-4 dimethylation (H3K4me2), H3K4me3, H3K9me2, H3K27me3 and H3K4 acetylation, but not for H3K9me3, 5-methylcytosine,or 5-hydroxymethylcytosine. At specific gene loci, the relative enrichments of histone and DNA modifications were changed to different degree in Hnf4a-deficient mouse liver. Among the epigenetic signatures investigated, changes in H3K4me3 correlated the best with mRNA expression. Additionally, Hnf4a-deficient livers had increased mRNA expression of histone H1.2 and H3.3 as well as epigenetic modifiers Dnmt1, Tet3, Setd7, Kmt2c, Ehmt2, and Ezh2. In conclusion, the present study provides convenient improved (h)MeDIP- and ChIP-qPCR assays for epigenetic study. Hnf4a deficiency in young-adult mouse liver markedly alters histone methylation and acetylation, with fewer effects on DNA methylation and 5-hydroxymethylation. The underlying mechanism may be the induction of epigenetic enzymes responsible for the addition/removal of the epigenetic signatures, and/or the loss of HNF4α per se as a key coordinator for epigenetic modifiers. | 24427299
|
Genome-wide evaluation of histone methylation changes associated with leaf senescence in Arabidopsis.
Brusslan, JA; Rus Alvarez-Canterbury, AM; Nair, NU; Rice, JC; Hitchler, MJ; Pellegrini, M
PloS one
7
e33151
2012
Show Abstract
Leaf senescence is the orderly dismantling of older tissue that allows recycling of nutrients to developing portions of the plant and is accompanied by major changes in gene expression. Histone modifications correlate to levels of gene expression, and this study utilizes ChIP-seq to classify activating H3K4me3 and silencing H3K27me3 marks on a genome-wide scale for soil-grown mature and naturally senescent Arabidopsis leaves. ChIPnorm was used to normalize data sets and identify genomic regions with significant differences in the two histone methylation patterns, and the differences were correlated to changes in gene expression. Genes that showed an increase in the H3K4me3 mark in older leaves were senescence up-regulated, while genes that showed a decrease in the H3K4me3 mark in the older leaves were senescence down-regulated. For the H3K27me3 modification, genes that lost the H3K27me3 mark in older tissue were senescence up-regulated. Only a small number of genes gained the H3K27me3 mark, and these were senescence down-regulated. Approximately 50% of senescence up-regulated genes lacked the H3K4me3 mark in both mature and senescent leaf tissue. Two of these genes, SAG12 and At1g73220, display strong senescence up-regulation without the activating H3K4me3 histone modification. This study provides an initial epigenetic framework for the developmental transition into senescence. | 22427974
|