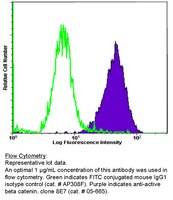
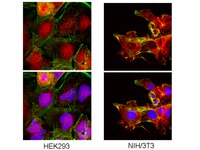
Paracrine factors from adipose-mesenchymal stem cells enhance metastatic capacity through Wnt signaling pathway in a colon cancer cell co-culture model.
Chen, D; Liu, S; Ma, H; Liang, X; Ma, H; Yan, X; Yang, B; Wei, J; Liu, X
Cancer cell international
15
42
2015
Show Abstract
Mesenchymal stem cells (MSCs) in tumors have emerged as progenitors involved in stroma formation and metastasis of cancers, partially owing to their abilities to differentially express paracrine factors related to the proliferation and invasion of cancer cells. In this regard, increasing evidence has shown that MSCs have impacts on the malignancy of colon cancer, however, the underpinning mechanisms by which MSCs promote cancer metastasis remain elusive.To investigate the crosstalk between adipose-derived MSCs (AMSCs) isolated from adipose tissues and colon cancer cells, a co-culture transwell model of AMSCs and colon cancer cells was employed, and the activation of Wnt signaling and paracrine factors in colon cancer cells and AMSCs were measured.The results showed that AMSCs could enhance the metastatic capacity of colon cancer cells with an elevated expression of mesenchymal-epithelial transition (EMT)-associated genes in a contact-dependent manner. Reciprocally, colon cancer cells were able to induce AMSCs to produce metastasis-related factors and cytokines, such as FGF10, VEGFC and matrix metalloproteinases (MMPs) in part through a mechanism of an activation of Wnt signaling, by which these factors in turn activate Wnt signaling of colon cancer cells. Intriguingly, an inhibition of Wnt signaling leads a reduced capacity of invasion and colony formation of colon cancer cells in vitro, and the tumorigenicity of cancer cells in a murine model.These findings thus suggest that the crosstalk between the Wnt signaling of cancer cells and paracrine factors of AMSCs has an implication in colon cancer malignancy. This study thus uncovers a novel Wnt-paracrine factors mediated-crosstalk between colon cancer cells and AMSCs in cancer malignancy. | | | 26060426
 |
Endocytic Adaptor Protein Tollip Inhibits Canonical Wnt Signaling.
Toruń, A; Szymańska, E; Castanon, I; Wolińska-Nizioł, L; Bartosik, A; Jastrzębski, K; Miętkowska, M; González-Gaitán, M; Miaczynska, M
PloS one
10
e0130818
2015
Show Abstract
Many adaptor proteins involved in endocytic cargo transport exhibit additional functions in other cellular processes which may be either related to or independent from their trafficking roles. The endosomal adaptor protein Tollip is an example of such a multitasking regulator, as it participates in trafficking and endosomal sorting of receptors, but also in interleukin/Toll/NF-κB signaling, bacterial entry, autophagic clearance of protein aggregates and regulation of sumoylation. Here we describe another role of Tollip in intracellular signaling. By performing a targeted RNAi screen of soluble endocytic proteins for their additional functions in canonical Wnt signaling, we identified Tollip as a potential negative regulator of this pathway in human cells. Depletion of Tollip potentiates the activity of β-catenin/TCF-dependent transcriptional reporter, while its overproduction inhibits the reporter activity and expression of Wnt target genes. These effects are independent of dynamin-mediated endocytosis, but require the ubiquitin-binding CUE domain of Tollip. In Wnt-stimulated cells, Tollip counteracts the activation of β-catenin and its nuclear accumulation, without affecting its total levels. Additionally, under conditions of ligand-independent signaling, Tollip inhibits the pathway after the stage of β-catenin stabilization, as observed in human cancer cell lines, characterized by constitutive β-catenin activity. Finally, the regulation of Wnt signaling by Tollip occurs also during early embryonic development of zebrafish. In summary, our data identify a novel function of Tollip in regulating the canonical Wnt pathway which is evolutionarily conserved between fish and humans. Tollip-mediated inhibition of Wnt signaling may contribute not only to embryonic development, but also to carcinogenesis. Mechanistically, Tollip can potentially coordinate multiple cellular pathways of trafficking and signaling, possibly by exploiting its ability to interact with ubiquitin and the sumoylation machinery. | | | 26110841
|
Slug-dependent upregulation of L1CAM is responsible for the increased invasion potential of pancreatic cancer cells following long-term 5-FU treatment.
Lund, K; Dembinski, JL; Solberg, N; Urbanucci, A; Mills, IG; Krauss, S
PloS one
10
e0123684
2015
Show Abstract
Pancreatic adenocarcinoma is a lethal disease with 5-year survival of less than 5%. 5-fluorouracil (5-FU) is a principal first-line therapy, but treatment only extends survival modestly and is seldom curative. Drug resistance and disease recurrence is typical and there is a pressing need to overcome this. To investigate acquired 5-FU resistance in pancreatic adenocarcinoma, we established chemoresistant monoclonal cell lines from the Panc 03.27 cell line by long-term exposure to increasing doses of 5-FU.5-FU-resistant cell lines exhibited increased expression of markers associated with multidrug resistance explaining their reduced sensitivity to 5-FU. In addition, 5-FU-resistant cell lines showed alterations typical for an epithelial-to-mesenchymal transition (EMT), including upregulation of mesenchymal markers and increased invasiveness. Microarray analysis revealed the L1CAM pathway as one of the most upregulated pathways in the chemoresistant clones, and a significant upregulation of L1CAM was seen on the RNA and protein level. In pancreatic cancer, expression of L1CAM is associated with a chemoresistant and migratory phenotype. Using esiRNA targeting L1CAM, or by blocking the extracellular part of L1CAM with antibodies, we show that the increased invasiveness observed in the chemoresistant cells functionally depends on L1CAM. Using esiRNA targeting β-catenin and/or Slug, we demonstrate that in the chemoresistant cell lines, L1CAM expression depends on Slug rather than β-catenin.Our findings establish Slug-induced L1CAM expression as a mediator of a chemoresistant and migratory phenotype in pancreatic adenocarcinoma cells. | | | 25860483
|
ChIP-Seq and RNA-Seq analyses identify components of the Wnt and Fgf signaling pathways as Prep1 target genes in mouse embryonic stem cells.
Laurent, A; Calabrese, M; Warnatz, HJ; Yaspo, ML; Tkachuk, V; Torres, M; Blasi, F; Penkov, D
PloS one
10
e0122518
2015
Show Abstract
The Prep1 (Pknox1) homeodomain transcription factor is essential at multiple stages of embryo development. In the E11.5 embryo trunk, we previously estimated that Prep1 binds about 3,300 genomic sites at a highly specific decameric consensus sequence, mainly governing basal cellular functions. We now show that in embryonic stem (ES) cells Prep1 binding pattern only partly overlaps that of the embryo trunk, with about 2,000 novel sites. Moreover, in ES cells Prep1 still binds mostly to promoters, as in total embryo trunk but, among the peaks bound exclusively in ES cells, the percentage of enhancers was three-fold higher. RNA-seq identifies about 1800 genes down-regulated in Prep1-/- ES cells which belong to gene ontology categories not enriched in the E11.5 Prep1i/i differentiated embryo, including in particular essential components of the Wnt and Fgf pathways. These data agree with aberrant Wnt and Fgf expression levels in the Prep1-/- ES cells with a deficient embryoid bodies (EBs) formation and differentiation. Re-establishment of the Prep1 level rescues the phenotype. | | | 25875616
|
β-Catenin and NF-κB co-activation triggered by TLR3 stimulation facilitates stem cell-like phenotypes in breast cancer.
Jia, D; Yang, W; Li, L; Liu, H; Tan, Y; Ooi, S; Chi, L; Filion, LG; Figeys, D; Wang, L
Cell death and differentiation
22
298-310
2015
Show Abstract
Cancer stem cells (CSCs) are responsible for tumor initiation and progression. Toll-like receptors (TLRs) are highly expressed in cancer cells and associated with poor prognosis. However, a linkage between CSCs and TLRs is unclear, and potential intervention strategies to prevent TLR stimulation-induced CSC formation and underlying mechanisms are lacking. Here, we demonstrate that stimulation of toll-like receptor 3 (TLR3) promotes breast cancer cells toward a CSC phenotype in vitro and in vivo. Importantly, conventional NF-κB signaling pathway is not exclusively responsible for TLR3 activation-enriched CSCs. Intriguingly, simultaneous activation of both β-catenin and NF-κB signaling pathways, but neither alone, is required for the enhanced CSC phenotypes. We have further identified a small molecule cardamonin that can concurrently inhibit β-catenin and NF-κB signals. Cardamonin is capable of effectively abolishing TLR3 activation-enhanced CSC phenotypes in vitro and successfully controlling TLR3 stimulation-induced tumor growth in human breast cancer xenografts. These findings may provide a foundation for developing new strategies to prevent the induction of CSCs during cancer therapies. | Western Blotting | | 25257174
|
Prostaglandin E2 promotes MYCN non-amplified neuroblastoma cell survival via β-catenin stabilization.
Jansen, SR; Holman, R; Hedemann, I; Frankes, E; Elzinga, CR; Timens, W; Gosens, R; de Bont, ES; Schmidt, M
Journal of cellular and molecular medicine
19
210-26
2015
Show Abstract
Amplification of MYCN is the most well-known prognostic marker of neuroblastoma risk classification, but still is only observed in 25% of cases. Recent evidence points to the cyclic adenosine monophosphate (cAMP) elevating ligand prostaglandin E2 (PGE2 ) and β-catenin as two novel players in neuroblastoma. Here, we aimed to define the potential role of PGE2 and cAMP and its potential interplay with β-catenin, both of which may converge on neuroblastoma cell behaviour. Gain and loss of β-catenin function, PGE2 , the adenylyl cyclase activator forskolin and pharmacological inhibition of cyclooxygenase-2 (COX-2) were studied in two human neuroblastoma cell lines without MYCN amplification. Our findings show that PGE2 enhanced cell viability through the EP4 receptor and cAMP elevation, whereas COX-2 inhibitors attenuated cell viability. Interestingly, PGE2 and forskolin promoted glycogen synthase kinase 3β inhibition, β-catenin phosphorylation at the protein kinase A target residue ser675, β-catenin nuclear translocation and TCF-dependent gene transcription. Ectopic expression of a degradation-resistant β-catenin mutant enhances neuroblastoma cell viability and inhibition of β-catenin with XAV939 prevented PGE2 -induced cell viability. Finally, we show increased β-catenin expression in human high-risk neuroblastoma tissue without MYCN amplification. Our data indicate that PGE2 enhances neuroblastoma cell viability, a process which may involve cAMP-mediated β-catenin stabilization, and suggest that this pathway is of relevance to high-risk neuroblastoma without MYCN amplification. | Western Blotting | Human | 25266063
|
Wnt-C59 arrests stemness and suppresses growth of nasopharyngeal carcinoma in mice by inhibiting the Wnt pathway in the tumor microenvironment.
Cheng, Y; Phoon, YP; Jin, X; Chong, SY; Ip, JC; Wong, BW; Lung, ML
Oncotarget
6
14428-39
2015
Show Abstract
Wnt/β-catenin signaling is responsible for the generation of cancer stem cells (CSCs) in many human tumors, including nasopharyngeal carcinoma (NPC). Recent studies demonstrate that Wnt or PORCN inhibitor, Wnt-C59, inhibits tumor growth in MMTV-WNT1 transgenic mice. The effect of Wnt-C59 in human tumors is not clear. In this study, the NPC cell lines investigated manifest heterogeneous responses to Wnt-C59 treatment. Wnt-C59 decreased tumor growth of SUNE1 cells in mice immediately following the administration of Wnt-C59. Mice injected with HNE1 cells did not develop visible tumors after the treatment of Wnt-C59, while control mice developed 100% tumors. Wnt-C59 inhibited stemness properties of NPC cells in a dosage-dependent manner by arresting sphere formation in both HNE1 and SUNE1 cells. Thus, Wnt-C59 has the potential to eradicate CSCs in human tumors. Active β-catenin and Axin2 proteins were strongly expressed in stromal cells surrounding growing tumors, confirming the importance of Wnt signaling activities in the microenvironment being driving forces for cell growth. These novel findings confirm the ability of Wnt-C59 to suppress Wnt-driven undifferentiated cell growth in NPC. Both anti-Wnt signaling and anti-CSC approaches are feasible strategies in cancer therapy. | | | 25980501
|
(Pro)renin receptor is crucial for Wnt/β-catenin-dependent genesis of pancreatic ductal adenocarcinoma.
Shibayama, Y; Fujimori, T; Nguyen, G; Hirose, T; Totsune, K; Ichihara, A; Kitada, K; Nakano, D; Kobori, H; Kohno, M; Masaki, T; Suzuki, Y; Yachida, S; Nishiyama, A
Scientific reports
5
8854
2015
Show Abstract
Although Wnt/β-catenin signaling is known to be aberrantly activated in PDAC, mutations of CTNNB1, APC or other pathway components are rare in this tumor type, suggesting alternative mechanisms for Wnt/β-catenin activation. Recent studies have implicated the (pro)renin receptor ((P)RR) is related to the Wnt/β-catenin signaling pathway. We therefore investigated the possible role of (P)RR in pancreatic carcinogenesis. Plasma s(P)RR levels were significantly (P less than 0.0001) higher in patients with PDAC than in healthy matched controls. We also identified aberrant expression of (P)RR in premalignant PanIN and PDAC lesions and all the PDAC cell lines examined. Inhibiting (P)RR with an siRNA attenuated activation of Wnt/β-catenin signaling pathway and reduced the proliferative ability of PDAC cells in vitro and the growth of engrafted tumors in vivo. Loss of (P)RR induced apoptosis of human PDAC cells. This is the first demonstration that (P)RR may be profoundly involved in ductal tumorigenesis in the pancreas. | | | 25747895
|
PBK/TOPK enhances aggressive phenotype in prostate cancer via β-catenin-TCF/LEF-mediated matrix metalloproteinases production and invasion.
Brown-Clay, JD; Shenoy, DN; Timofeeva, O; Kallakury, BV; Nandi, AK; Banerjee, PP
Oncotarget
6
15594-609
2015
Show Abstract
A current challenge in prostate cancer treatment is how to differentiate aggressive disease from indolent prostate cancer. There is an urgent need to identify markers that would accurately distinguish indolent prostate cancer from aggressive disease. The aim of this study was to evaluate the role of PDZ Domain-binding kinase (PBK) in prostate cancer and to determine if PBK expression enhances aggressiveness in prostate cancer. Using archival tissue samples, gain-of-function and loss-of-function studies, we show that PBK expression is up-regulated in prostate cancer, and its expression level is commensurate with invasiveness. Modulation of PBK expression and function causally regulates the invasive ability of prostate cancer cells. Production of matrix metalloproteinases-2 and -9, which are key players in metastatic invasion, is up-regulated, and the promoters of these genes are transcriptionally activated by PBK via increased β-catenin-TCF/LEF signaling. Prostate cancer tissue specimens show that PBK's expression correlates with aggressive disease and distant metastasis in bone, lymph node and abdomen. Our in vitro and in situ data are in agreement that PBK could be a prognostic biomarker for prostate cancer that would discriminate aggressive prostate cancer from indolent disease, and is a potential target for the therapeutic intervention of aggressive prostate cancer in men. | | | 25909225
|
Mechanism of Resistance and Novel Targets Mediating Resistance to EGFR and c-Met Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer.
Botting, GM; Rastogi, I; Chhabra, G; Nlend, M; Puri, N
PloS one
10
e0136155
2015
Show Abstract
Tyrosine kinase inhibitors (TKIs) against EGFR and c-Met are initially effective when administered individually or in combination to non-small cell lung cancer (NSCLC) patients. However, the overall efficacies of TKIs are limited due to the development of drug resistance. Therefore, it is important to elucidate mechanisms of EGFR and c-Met TKI resistance in order to develop more effective therapies. Model NSCLC cell lines H1975 and H2170 were used to study the similarities and differences in mechanisms of EGFR/c-Met TKI resistance. H1975 cells are positive for the T790M EGFR mutation, which confers resistance to current EGFR TKI therapies, while H2170 cells are EGFR wild-type. Previously, H2170 cells were made resistant to the EGFR TKI erlotinib and the c-Met TKI SU11274 by exposure to progressively increasing concentrations of TKIs. In H2170 and H1975 TKI-resistant cells, key Wnt and mTOR proteins were found to be differentially modulated. Wnt signaling transducer, active β-catenin was upregulated in TKI-resistant H2170 cells when compared to parental cells. GATA-6, a transcriptional activator of Wnt, was also found to be upregulated in resistant H2170 cells. In H2170 erlotinib resistant cells, upregulation of inactive GSK3β (p-GSK3β) was observed, indicating activation of Wnt and mTOR pathways which are otherwise inhibited by its active form. However, in H1975 cells, Wnt modulators such as active β-catenin, GATA-6 and p-GSK3β were downregulated. Additional results from MTT cell viability assays demonstrated that H1975 cell proliferation was not significantly decreased after Wnt inhibition by XAV939, but combination treatment with everolimus (mTOR inhibitor) and erlotinib resulted in synergistic cell growth inhibition. Thus, in H2170 cells and H1975 cells, simultaneous inhibition of key Wnt or mTOR pathway proteins in addition to EGFR and c-Met may be a promising strategy for overcoming EGFR and c-Met TKI resistance in NSCLC patients. | | | 26301867
|