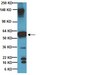
A novel approach to preventing the hemolysis of paroxysmal nocturnal hemoglobinuria: both complement-mediated cytolysis and C3 deposition are blocked by a monoclonal antibody specific for the alternative pathway of complement.
Lindorfer, MA; Pawluczkowycz, AW; Peek, EM; Hickman, K; Taylor, RP; Parker, CJ
Blood
115
2283-91
2010
Show Abstract
The clinical hallmark of paroxysmal nocturnal hemoglobinuria (PNH) is chronic intravascular hemolysis that is a consequence of unregulated activation of the alternative pathway of complement (APC). Intravascular hemolysis can be inhibited in patients by treatment with eculizumab, a monoclonal antibody that binds complement C5 thereby preventing formation of the cytolytic membrane attack complex of complement. However, in essentially all patients treated with eculizumab, persistent anemia, reticulocytosis, and biochemical evidence of hemolysis are observed; and in a significant proportion, their PNH erythrocytes become opsonized with complement C3. These observations suggest that PNH patients treated with eculizumab are left with clinically significant immune-mediated hemolytic anemia because the antibody does not block APC activation. With a goal of improving PNH therapy, we characterized the activity of anti-C3b/iC3b monoclonal antibody 3E7 in an in vitro model of APC-mediated hemolysis. We show that 3E7 and its chimeric-deimmunized derivative H17 block both hemolysis and C3 deposition on PNH erythrocytes. The antibody is specific for the APC C3/C5 convertase because classical pathway-mediated hemolysis is unaffected by 3E7/H17. These findings suggest an approach to PNH treatment in which both intravascular and extravascular hemolysis can be inhibited while preserving important immune functions of the classical pathway of complement. | 20068220
 |
An anti-C3b(i) mAb enhances complement activation, C3b(i) deposition, and killing of CD20+ cells by rituximab.
Kennedy, AD; Solga, MD; Schuman, TA; Chi, AW; Lindorfer, MA; Sutherland, WM; Foley, PL; Taylor, RP
Blood
101
1071-9
2003
Show Abstract
We investigated deposition of the complement protein fragment C3b and its breakdown products (collectively designated as C3b(i)) on CD20-positive cells treated with rituximab (RTX) in the presence of normal human serum (NHS). Radioimmunoassay (RIA) demonstrates that about 500 000 C3b(i) molecules deposit per cell, and fluorescence microscopy reveals that C3b(i) colocalizes with bound RTX. Use of mAb 3E7, specific for C3b(i) bound to substrates, enhances C3b(i) deposition; > 1 million C3b(i) deposit when cells are incubated with NHS, RTX and mAb 3E7. Treatment of Raji cells in NHS plus RTX leads to robust cell killing (95%) after 24 to 48 hours, and mAb 3E7 significantly enhances RTX-mediated killing of Raji and DB cells. A cynomolgus monkey model based on intravenous infusion of RTX followed by mAb 3E7 demonstrated that RTX rapidly binds to B cells and promotes complement activation and C3b(i) deposition; fluorescence microscopy analyses revealed the same pattern of colocalization of C3b(i) on cell-bound RTX in vivo as observed in vitro. Preliminary in vitro studies with blood samples from patients with chronic lymphocytic leukemia lead to similar findings. These experiments suggest that complement plays a key role in the mechanism of action of RTX; moreover, the in vivo molecular form of RTX (and possibly other antitumor mAbs) in the circulation or in tissues may include C3b(i) molecules covalently bound to the therapeutic mAb, thus allowing it to interact with cells containing both Fc and complement receptors. | 12393727
|