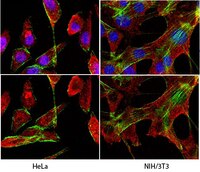
Connexin43 Mediated Delivery of ADAMTS5 Targeting siRNAs from Mesenchymal Stem Cells to Synovial Fibroblasts.
Liu, S; Niger, C; Koh, EY; Stains, JP
PloS one
10
e0129999
2015
Show Abstract
Osteoarthritis is a joint-destructive disease that has no effective cure. Human mesenchymal stem cells (hMSCs) could offer therapeutic benefit in the treatment of arthritic diseases by suppressing inflammation and permitting tissue regeneration, but first these cells must overcome the catabolic environment of the diseased joint. Likewise, gene therapy also offers therapeutic promise given its ability to directly modulate key catabolic factors that mediate joint deterioration, although it too has limitations. In the current study, we explore an approach that combines hMSCs and gene therapy. Specifically, we test the use of hMSC as a vehicle to deliver ADAMTS5 (an aggrecanase with a key role in osteoarthritis)-targeting siRNAs to SW982 synovial fibroblast-like cells via connexin43 containing gap junctions. Accordingly, we transduced hMSCs with ADAMTS5-targeting shRNA or non-targeted shRNA, and co-cultured them with synovial fibroblasts to allow delivery of siRNAs from hMSC to synovial fibroblasts. We found that co-culture of hMSCs-shRNA-ADAMTS5 and synovial fibroblasts reduced ADAMTS5 expression relative to co-culture of hMSCs-shRNA-control and synovial fibroblasts. Furthermore, ADAMTS5 was specifically reduced in the synovial fibroblasts populations as determined by fluorescence-activated cell sorting, suggesting transfer of the siRNA between cells. To test if Cx43-containing gap junctions are involved in the transfer of siRNA, we co-cultured hMSCs-shRNA-ADAMTS5 cells with synovial fibroblasts in which connexin43 was knocked down. Under these conditions, ADAMTS5 levels were not inhibited by co-culture, indicating that connexin43 mediates the delivery of siRNA from hMSCs to synovial fibroblasts. In total, our findings demonstrate that hMSCs can function as donor cells to host and deliver siRNAs to synovial fibroblasts via connexin43 gap junction in vitro. These data may have implications in the combination of hMSCs and gene therapy to treat diseases like osteoarthritis, in vivo. | | | 26076025
 |
Fibroblast-epithelial cell interactions drive epithelial-mesenchymal transition differently in cells from normal and COPD patients.
Nishioka, M; Venkatesan, N; Dessalle, K; Mogas, A; Kyoh, S; Lin, TY; Nair, P; Baglole, CJ; Eidelman, DH; Ludwig, MS; Hamid, Q
Respiratory research
16
72
2015
Show Abstract
Epithelial-to-mesenchymal transition (EMT), which involves changes in cellular morphology of highly polarized epithelial cells and the gain of mesenchymal cell phenotype with migratory and invasive capacities, is implicated in smoking-related chronic obstructive pulmonary disease (COPD). However, the interactions of fibroblasts and epithelial cells and the participation of fibroblasts in the EMT processes in COPD are poorly understood. Here, we investigated the hypothesis that EMT is active in human bronchial epithelial (HBE) cells of COPD patients, and that mediators secreted by lung fibroblasts from COPD patients induce EMT.Primary HBE cells from normal subjects and COPD patients were purchased from LONZA. HLFs were derived from resected lung obtained from normal (N) and COPD (D) subjects and their conditioned medium (CM) was collected after 2-day culture in serum-free medium. The expression of epithelial and mesenchymal markers as well as EMT-related transcription factors in lung biopsies, and in HBE cells following stimulation with CM from both normal human lung fibroblasts (NHLF) and COPD human lung fibroblasts (DHLF) was evaluated by immunohistochemistry, qRT-PCR and western blot.Basal mRNA expression of mesenchymal markers and EMT-related transcription factors were increased in DHBE cells compared to normal human bronchial epithelial cells (NHBE) cells as well as in COPD lungs. CM from NHLF significantly induced vimentin expression in both NHBE and COPD human bronchial epithelial cells (DHBE) cells, but only increased N-cadherin expression in DHBE cells. CM from NHLF significantly induced Twist1 and Twist2 expression in NHBE cells and increased Snai2 (Slug) expression in DHBE cells. While CM from NHLF had no effect on such EMT markers, CM from DHLF significantly increased the protein expression of E-cadherin and vimentin in NHBE cells compared to control. N-cadherin expression was upregulated to a greater degree in NHBE cells than DHBE cells. Only CM from DHLF significantly increased E-/N-cadherin ratio in DHBE cells.Our results suggest that DHBE cells have partially undergone EMT under baseline conditions. DHLF-CM promoted EMT in NHBE, suggesting that interactions between fibroblast and epithelial cells may play an important role in the EMT process in COPD. | | | 26081431
|
Inhibition of ERBB2-overexpressing Tumors by Recombinant Human Prolidase and Its Enzymatically Inactive Mutant.
Yang, L; Li, Y; Bhattacharya, A; Zhang, Y
EBioMedicine
2
396-405
2015
Show Abstract
ERBB2 is an oncogenic receptor tyrosine kinase overexpressed in a subset of human breast cancer and other cancers. We recently found that human prolidase (PEPD), a dipeptidase, is a high affinity ERBB2 ligand and cross-links two ERBB2 monomers. Here, we show that recombinant human PEPD (rhPEPD) strongly inhibits ERBB2-overexpressing tumors in mice, whereas it does not impact tumors without ERBB2 overexpression. rhPEPD causes ERBB2 depletion, disrupts oncogenic signaling orchestrated by ERBB2 homodimers and heterodimers, and induces apoptosis. The impact of enzymatically-inactive mutant rhPEPD(G278D) on ERBB2 is indistinguishable from that of rhPEPD, but rhPEPD(G278D) is superior to rhPEPD for tumor inhibition. The enzymatic function of rhPEPD stimulates HIF-1α and other pro-survival factors in tumors, which likely attenuates its antitumor activity. rhPEPD(G278D) is also attractive in that it may not interfere with the physiologic function of endogenous PEPD in normal cells. Collectively, we have identified a human protein as an inhibitory ERBB2 ligand that inhibits ERBB2-overexpressing tumors in vivo. Several anti-ERBB2 agents are on the market but are hampered by drug resistance and high drug cost. rhPEPD(G278D) may synergize with these agents and may also be highly cost-effective, since it targets ERBB2 with a different mechanism and can be produced in bacteria. | | | 26086037
|
MURC/cavin-4 Is Co-Expressed with Caveolin-3 in Rhabdomyosarcoma Tumors and Its Silencing Prevents Myogenic Differentiation in the Human Embryonal RD Cell Line.
Faggi, F; Codenotti, S; Poliani, PL; Cominelli, M; Chiarelli, N; Colombi, M; Vezzoli, M; Monti, E; Bono, F; Tulipano, G; Fiorentini, C; Zanola, A; Lo, HP; Parton, RG; Keller, C; Fanzani, A
PloS one
10
e0130287
2015
Show Abstract
The purpose of this study was to investigate whether MURC/cavin-4, a plasma membrane and Z-line associated protein exhibiting an overlapping distribution with Caveolin-3 (Cav-3) in heart and muscle tissues, may be expressed and play a role in rhabdomyosarcoma (RMS), an aggressive myogenic tumor affecting childhood. We found MURC/cavin-4 to be expressed, often concurrently with Cav-3, in mouse and human RMS, as demonstrated through in silico analysis of gene datasets and immunohistochemical analysis of tumor samples. In vitro expression studies carried out using human cell lines and primary mouse tumor cultures showed that expression levels of both MURC/cavin-4 and Cav-3, while being low or undetectable during cell proliferation, became robustly increased during myogenic differentiation, as detected via semi-quantitative RT-PCR and immunoblotting analysis. Furthermore, confocal microscopy analysis performed on human RD and RH30 cell lines confirmed that MURC/cavin-4 mostly marks differentiated cell elements, colocalizing at the cell surface with Cav-3 and labeling myosin heavy chain (MHC) expressing cells. Finally, MURC/cavin-4 silencing prevented the differentiation in the RD cell line, leading to morphological cell impairment characterized by depletion of myogenin, Cav-3 and MHC protein levels. Overall, our data suggest that MURC/cavin-4, especially in combination with Cav-3, may play a consistent role in the differentiation process of RMS. | | | 26086601
|
Dendritic Cell-Mediated Phagocytosis but Not Immune Activation Is Enhanced by Plasmin.
Borg, RJ; Samson, AL; Au, AE; Scholzen, A; Fuchsberger, M; Kong, YY; Freeman, R; Mifsud, NA; Plebanski, M; Medcalf, RL
PloS one
10
e0131216
2015
Show Abstract
Removal of dead cells in the absence of concomitant immune stimulation is essential for tissue homeostasis. We recently identified an injury-induced protein misfolding event that orchestrates the plasmin-dependent proteolytic degradation of necrotic cells. As impaired clearance of dead cells by the innate immune system predisposes to autoimmunity, we determined whether plasmin could influence endocytosis and immune cell stimulation by dendritic cells - a critical cell that links the innate and adaptive immune systems. We find that plasmin generated on the surface of necrotic cells enhances their phagocytic removal by human monocyte-derived dendritic cells. Plasmin also promoted phagocytosis of protease-resistant microparticles by diverse mouse dendritic cell sub-types both in vitro and in vivo. Together with an increased phagocytic capacity, plasmin-treated dendritic cells maintain an immature phenotype, exhibit reduced migration to lymph nodes, increase their expression/release of the immunosuppressive cytokine TGF-β, and lose their capacity to mount an allogeneic response. Collectively, our findings support a novel role for plasmin formed on dead cells and other phagocytic targets in maintaining tissue homeostasis by increasing the phagocytic function of dendritic cells while simultaneously decreasing their immunostimulatory capacity consistent with producing an immunosuppressive state. | | | 26132730
|
TGF-Beta Blockade Increases Renal Inflammation Caused by the C-Terminal Module of the CCN2.
Rodrigues-Díez, R; Rayego-Mateos, S; Orejudo, M; Aroeira, LS; Selgas, R; Ortiz, A; Egido, J; Ruiz-Ortega, M
Mediators of inflammation
2015
506041
2015
Show Abstract
The CCN family member 2 (CCN2, also known as connective tissue growth factor) may behave as a risk biomarker and a potential therapeutic target for renal disease. CCN2 participates in the regulation of inflammation and fibrosis. TGF-β is considered the main fibrogenic cytokine; however, in some pathological settings TGF-β also has anti-inflammatory properties. CCN2 has been proposed as a downstream profibrotic mediator of TGF-β, but data on TGF-β role in CCN2 actions are scarce. Our aim was to evaluate the effect of TGF-β blockade in CCN2-mediated experimental renal damage. Systemic administration of the C-terminal module of CCN2 to mice caused sustained renal inflammation. In these mice, TGF-β blockade, using an anti-TGF-β neutralizing antibody, significantly increased renal expression of the NGAL (a kidney injury biomarker), kidney infiltration by monocytes/macrophages, and upregulation of MCP-1 expression. The anti-inflammatory effect of TGF-β seems to be mediated by a dysregulation of the systemic Treg immune response, shown by decreased levels of circulating CD4(+)/Foxp3(+)Treg cells. Our experimental data support the idea that TGF-β exerts anti-inflammatory actions in the kidney and suggest that it is not an optimal therapeutic target. | | | 26074680
|
Huntington disease iPSCs show early molecular changes in intracellular signaling, the expression of oxidative stress proteins and the p53 pathway.
Szlachcic, WJ; Switonski, PM; Krzyzosiak, WJ; Figlerowicz, M; Figiel, M
Disease models & mechanisms
8
1047-57
2015
Show Abstract
Huntington disease (HD) is a brain disorder characterized by the late onset of motor and cognitive symptoms, even though the neurons in the brain begin to suffer dysfunction and degeneration long before symptoms appear. There is currently no cure. Several molecular and developmental effects of HD have been identified using neural stem cells (NSCs) and differentiated cells, such as neurons and astrocytes. Still, little is known regarding the molecular pathogenesis of HD in pluripotent cells, such as embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). Therefore, we examined putative signaling pathways and processes involved in HD pathogenesis in pluripotent cells. We tested naïve mouse HD YAC128 iPSCs and two types of human HD iPSC that were generated from HD and juvenile-HD patients. Surprisingly, we found that a number of changes affecting cellular processes in HD were also present in undifferentiated pluripotent HD iPSCs, including the dysregulation of the MAPK and Wnt signaling pathways and the dysregulation of the expression of genes related to oxidative stress, such as Sod1. Interestingly, a common protein interactor of the huntingtin protein and the proteins in the above pathways is p53, and the expression of p53 was dysregulated in HD YAC128 iPSCs and human HD iPSCs. In summary, our findings demonstrate that multiple molecular pathways that are characteristically dysregulated in HD are already altered in undifferentiated pluripotent cells and that the pathogenesis of HD might begin during the early stages of life. | Western Blotting | | 26092128
|
Pathophysiological role of microRNA-29 in pancreatic cancer stroma.
Kwon, JJ; Nabinger, SC; Vega, Z; Sahu, SS; Alluri, RK; Abdul-Sater, Z; Yu, Z; Gore, J; Nalepa, G; Saxena, R; Korc, M; Kota, J
Scientific reports
5
11450
2015
Show Abstract
Dense fibrotic stroma associated with pancreatic ductal adenocarcinoma (PDAC) is a major obstacle for drug delivery to the tumor bed and plays a crucial role in pancreatic cancer progression. Current, anti-stromal therapies have failed to improve tumor response to chemotherapy and patient survival. Furthermore, recent studies show that stroma impedes tumor progression, and its complete ablation accelerates PDAC progression. In an effort to understand the molecular mechanisms associated with tumor-stromal interactions, using in vitro and in vivo models and PDAC patient biopsies, we show that the loss of miR-29 is a common phenomenon of activated pancreatic stellate cells (PSCs)/fibroblasts, the major stromal cells responsible for fibrotic stromal reaction. Loss of miR-29 is correlated with a significant increase in extracellular matrix (ECM) deposition, a major component in PDAC stroma. Our in vitro miR-29 gain/loss-of-function studies document the role of miR-29 in PSC-mediated ECM stromal protein accumulation. Overexpression of miR-29 in activated stellate cells reduced stromal deposition, cancer cell viability, and cancer growth in co-culture. Furthermore, the loss of miR-29 in TGF-β1 activated PSCs is SMAD3 dependent. These results provide insights into the mechanistic role of miR-29 in PDAC stroma and its potential use as a therapeutic agent to target PDAC. | | | 26095125
|
A plastic relationship between vinculin-mediated tension and adhesion complex area defines adhesion size and lifetime.
Hernández-Varas, P; Berge, U; Lock, JG; Strömblad, S
Nature communications
6
7524
2015
Show Abstract
Cell-matrix adhesions are central mediators of mechanotransduction, yet the interplay between force and adhesion regulation remains unclear. Here we use live cell imaging to map time-dependent cross-correlations between vinculin-mediated tension and adhesion complex area, revealing a plastic, context-dependent relationship. Interestingly, while an expected positive cross-correlation dominated in mid-sized adhesions, small and large adhesions display negative cross-correlation. Furthermore, although large changes in adhesion complex area follow vinculin-mediated tension alterations, small increases in area precede vinculin-mediated tension dynamics. Modelling based on this mapping of the vinculin-mediated tension-adhesion complex area relationship confirms its biological validity, and indicates that this relationship explains adhesion size and lifetime limits, keeping adhesions focal and transient. We also identify a subpopulation of steady-state adhesions whose size and vinculin-mediated tension become stabilized, and whose disassembly may be selectively microtubule-mediated. In conclusion, we define a plastic relationship between vinculin-mediated tension and adhesion complex area that controls fundamental cell-matrix adhesion properties. | | | 26109125
|
Reliable LC3 and p62 autophagy marker detection in formalin fixed paraffin embedded human tissue by immunohistochemistry.
Schläfli, AM; Berezowska, S; Adams, O; Langer, R; Tschan, MP
European journal of histochemistry : EJH
59
2481
2015
Show Abstract
Autophagy assures cellular homeostasis, and gains increasing importance in cancer, where it impacts on carcinogenesis, propagation of the malignant phenotype and development of resistance. To date, its tissue-based analysis by immunohistochemistry remains poorly standardized. Here we show the feasibility of specifically and reliably assessing the autophagy markers LC3B and p62 (SQSTM1) in formalin fixed and paraffin embedded human tissue by immunohistochemistry. Preceding functional experiments consisted of depleting LC3B and p62 in H1299 lung cancer cells with subsequent induction of autophagy. Western blot and immunofluorescence validated antibody specificity, knockdown efficiency and autophagy induction prior to fixation in formalin and embedding in paraffin. LC3B and p62 antibodies were validated on formalin fixed and paraffin embedded cell pellets of treated and control cells and finally applied on a tissue microarray with 80 human malignant and non-neoplastic lung and stomach formalin fixed and paraffin embedded tissue samples. Dot-like staining of various degrees was observed in cell pellets and 18/40 (LC3B) and 22/40 (p62) tumors, respectively. Seventeen tumors were double positive for LC3B and p62. P62 displayed additional significant cytoplasmic and nuclear staining of unknown significance. Interobserver-agreement for grading of staining intensities and patterns was substantial to excellent (kappa values 0.60 - 0.83). In summary, we present a specific and reliable IHC staining of LC3B and p62 on formalin fixed and paraffin embedded human tissue. Our presented protocol is designed to aid reliable investigation of dysregulated autophagy in solid tumors and may be used on large tissue collectives. | | | 26150155
|