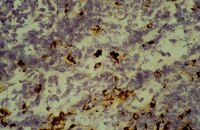
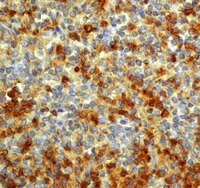
Interleukin-18 expression increases in response to neurovascular damage following soman-induced status epilepticus in rats.
Johnson, EA; Guignet, MA; Dao, TL; Hamilton, TA; Kan, RK
Journal of inflammation (London, England)
12
43
2015
Show Abstract
Status epilepticus (SE) can cause neuronal cell death and impaired behavioral function. Acute exposure to potent acetylcholinesterase inhibitors such as soman (GD) can cause prolonged SE activity, micro-hemorrhage and cell death in the hippocampus, thalamus and piriform cortex. Neuroinflammation is a prominent feature of brain injury with upregulation of multiple pro-inflammatory cytokines including those of the IL-1 family. The highly pleiotropic pro-inflammatory cytokine interleukin-18 (IL-18) belongs to the IL-1 family of cytokines and can propagate neuroinflammation by promoting immune cell infiltration, leukocyte and lymphocyte activation, and angiogenesis and helps facilitate the transition from the innate to the adaptive immune response. The purpose of this study is to characterize the regional and temporal expression of IL -18 and related factors in the brain following SE in a rat GD seizure model followed by localization of IL-18 to specific cell types.The protein levels of IL-18, vascular endothelial growth factor and interferon gamma was quantified in the lysates of injured brain regions up to 72 h following GD-induced SE onset using bead multiplex immunoassays. IL-18 was localized to various cell types using immunohistochemistry and transmission electron microscopy. In addition, macrophage appearance scoring and T-cell quantification was determined using immunohistochemistry. Micro-hemorrhages were identified using hematoxylin and eosin staining of brain sections.Significant increases in IL-18 occurred in the piriform cortex, hippocampus and thalamus following SE. IL-18 was primarily expressed by endothelial cells and astrocytes associated with the damaged neurovascular unit. The increase in IL-18 was not related to macrophage accumulation, neutrophil infiltration or T-cell appearance in the injured tissue.These data show that IL-18 is significantly upregulated following GD-induced SE and localized primarily to endothelial cells in damaged brain vasculature. IL-18 upregulation occurred following leukocyte/lymphocyte infiltration and in the absence of other IL-18-related cytokines, suggesting another function, potentially for angiogenesis related to GD-induced micro-hemorrhage formation. Further studies at more chronic time points may help to elucidate this function. | | 26203299
 |
Neuregulin-1 inhibits neuroinflammatory responses in a rat model of organophosphate-nerve agent-induced delayed neuronal injury.
Li, Y; Lein, PJ; Ford, GD; Liu, C; Stovall, KC; White, TE; Bruun, DA; Tewolde, T; Gates, AS; Distel, TJ; Surles-Zeigler, MC; Ford, BD
Journal of neuroinflammation
12
64
2015
Show Abstract
Neuregulin-1 (NRG-1) has been shown to act as a neuroprotectant in animal models of nerve agent intoxication and other acute brain injuries. We recently demonstrated that NRG-1 blocked delayed neuronal death in rats intoxicated with the organophosphate (OP) neurotoxin diisopropylflurophosphate (DFP). It has been proposed that inflammatory mediators are involved in the pathogenesis of OP neurotoxin-mediated brain damage.We examined the influence of NRG-1 on inflammatory responses in the rat brain following DFP intoxication. Microglial activation was determined by immunohistchemistry using anti-CD11b and anti-ED1 antibodies. Gene expression profiling was performed with brain tissues using Affymetrix gene arrays and analyzed using the Ingenuity Pathway Analysis software. Cytokine mRNA levels following DFP and NRG-1 treatment was validated by real-time reverse transcription polymerase chain reaction (RT-PCR).DFP administration resulted in microglial activation in multiple brain regions, and this response was suppressed by treatment with NRG-1. Using microarray gene expression profiling, we observed that DFP increased mRNA levels of approximately 1,300 genes in the hippocampus 24 h after administration. NRG-1 treatment suppressed by 50% or more a small fraction of DFP-induced genes, which were primarily associated with inflammatory responses. Real-time RT-PCR confirmed that the mRNAs for pro-inflammatory cytokines interleukin-1β (IL-1β) and interleukin-6 (IL-6) were significantly increased following DFP exposure and that NRG-1 significantly attenuated this transcriptional response. In contrast, tumor necrosis factor α (TNFα) transcript levels were unchanged in both DFP and DFP + NRG-1 treated brains relative to controls.Neuroprotection by NRG-1 against OP neurotoxicity is associated with the suppression of pro-inflammatory responses in brain microglia. These findings provide new insight regarding the molecular mechanisms involved in the neuroprotective role of NRG-1 in acute brain injuries. | | 25880399
|
Involvement of phosphatase and tensin homolog deleted from chromosome 10 in rodent model of neuropathic pain.
Huang, SY; Sung, CS; Chen, WF; Chen, CH; Feng, CW; Yang, SN; Hung, HC; Chen, NF; Lin, PR; Chen, SC; Wang, HM; Chu, TH; Tai, MH; Wen, ZH
Journal of neuroinflammation
12
59
2015
Show Abstract
Many cancer research studies have extensively examined the phosphatase and tensin homolog deleted from chromosome 10 (PTEN) pathway. There are only few reports that suggest that PTEN might affect pain; however, there is still a lack of evidence to show the role of PTEN for modulating pain. Here, we report a role for PTEN in a rodent model of neuropathic pain.We found that chronic constriction injury (CCI) surgery in rats could elicit downregulation of spinal PTEN as well as upregulation of phosphorylated PTEN (phospho-PTEN) and phosphorylated mammalian target of rapamycin (phospho-mTOR). After examining such changes in endogenous PTEN in neuropathic rats, we explored the effects of modulating the spinal PTEN pathway on nociceptive behaviors. The normal rats exhibited mechanical allodynia after intrathecal (i.t.) injection of adenovirus-mediated PTEN antisense oligonucleotide (Ad-antisense PTEN). These data indicate the importance of downregulation of spinal PTEN for nociception. Moreover, upregulation of spinal PTEN by i.t. adenovirus-mediated PTEN (Ad-PTEN) significantly prevented CCI-induced development of nociceptive sensitization, thermal hyperalgesia, mechanical allodynia, cold allodynia, and weight-bearing deficits in neuropathic rats. Furthermore, upregulation of spinal PTEN by i.t. Ad-PTEN significantly attenuated CCI-induced microglia and astrocyte activation, upregulation of tumor necrosis factor-α (TNF-α) and phospho-mTOR, and downregulation of PTEN in neuropathic rats 14 days post injury.These findings demonstrate that PTEN plays a key, beneficial role in a rodent model of neuropathic pain. | | 25889774
|
Neurochemical properties of BDNF-containing neurons projecting to rostral ventromedial medulla in the ventrolateral periaqueductal gray.
Yin, JB; Wu, HH; Dong, YL; Zhang, T; Wang, J; Zhang, Y; Wei, YY; Lu, YC; Wu, SX; Wang, W; Li, YQ
Frontiers in neural circuits
8
137
2014
Show Abstract
The periaqueductal gray (PAG) modulates nociception via a descending pathway that relays in the rostral ventromedial medulla (RVM) and terminates in the spinal cord. Previous behavioral pharmacology and electrophysiological evidence suggests that brain-derived neurotrophic factor (BDNF) plays an important role in descending pain modulation, likely through the PAG-RVM pathway. However, detailed information is still lacking on the distribution of BDNF, activation of BDNF-containing neurons projecting to RVM in the condition of pain, and neurochemical properties of these neurons within the PAG. Through fluorescent in situ hybridization (FISH) and immunofluorescent staining, the homogenous distributions of BDNF mRNA and protein were observed in the four subregions of PAG. Both neurons and astrocytes expressed BDNF, but not microglia. By combining retrograde tracing methods and formalin pain model, there were more BDNF-containing neurons projecting to RVM being activated in the ventrolateral subregion of PAG (vlPAG) than other subregions of PAG. The neurochemical properties of BDNF-containing projection neurons in the vlPAG were investigated. BDNF-containing projection neurons expressed the autoreceptor TrkB in addition to serotonin (5-HT), neurotensin (NT), substance P (SP), calcitonin gene related peptide (CGRP), nitric oxide synthase (NOS), and parvalbumin (PV) but not tyrosine decarboxylase (TH). It is speculated that BDNF released from projection neurons in the vlPAG might participate in the descending pain modulation through enhancing the presynaptic release of other neuroactive substances (NSs) in the RVM. | | 25477786
|
Suppression of spinal connexin 43 expression attenuates mechanical hypersensitivity in rats after an L5 spinal nerve injury.
Xu, Q; Cheong, YK; He, SQ; Tiwari, V; Liu, J; Wang, Y; Raja, SN; Li, J; Guan, Y; Li, W
Neuroscience letters
566
194-9
2014
Show Abstract
Activation of spinal astrocytes may contribute to neuropathic pain. Adjacent astrocytes can make direct communication through gap junctions formed by connexin 43 (Cx43) in the central nervous system. Yet, the role of spinal astroglial gap junctions in neuropathic pain is not fully understood. Since Cx43 is the connexin isoform expressed preferentially in astrocytes in the spinal cord, we used a small interfering RNA (siRNA) approach to examine whether suppression of spinal Cx43 expression inhibits mechanical hypersensitivity in rats after an L5 spinal nerve ligation (SNL). SNL rats were administered intrathecal Cx43 siRNA (3μg/15μl, twice/day) or an equal amount of mismatch siRNA (control) on days 14-17 post-SNL. Cx43 siRNA, but not mismatch siRNA, alleviated mechanical hypersensitivity in SNL rats. Furthermore, Western blot analysis showed that the pain inhibition induced by Cx43 siRNA correlated with downregulation of Cx43 expression, but not that of Cx36 (the neuronal gap junction protein) or glial fibrillary acidic protein (GFAP, a marker for reactive astrocytes) in the spinal cord of SNL rats. Western blot analysis and immunohistochemistry also showed that SNL increased GFAP expression, but decreased Cx43 expression, in spinal cord. Our results provide direct evidence that selective suppression of spinal Cx43 after nerve injury alleviates neuropathic mechanical hypersensitivity. These findings suggest that in the spinal cord, the enhanced function of astroglial gap junctions, especially those formed by Cx43, may be important to neuropathic pain in SNL rats. | | 24631560
|
Runx1t1 (Runt-related transcription factor 1; translocated to, 1) epigenetically regulates the proliferation and nitric oxide production of microglia.
Baby, N; Li, Y; Ling, EA; Lu, J; Dheen, ST
PloS one
9
e89326
2014
Show Abstract
Microglia, the resident immune cells of the brain, undergo rapid proliferation and produce several proinflammatory molecules and nitric oxide (NO) when activated in neuropathological conditions. Runx1t1 (Runt-related transcription factor 1, translocated to 1) has been implicated in recruiting histone deacetylases (HDACs) for transcriptional repression, thereby regulating cell proliferation. In the present study, Runx1t1 expression was shown to localize in amoeboid microglial cells of the postnatal rat brain, being hardly detectable in ramified microglia of the adult brain. Moreover, a marked expression of Runx1t1was induced and translocated to nuclei in activated microglia in vitro and in vivo. In view of these findings, it was hypothesized that Runx1t1 regulates microglial functions during development and in neuropathological conditions.siRNA-mediated knockdown of Runx1t1 significantly decreased the expression level of cell cycle-related gene, cyclin-dependent kinase 4 (Cdk4) and proliferation index in activated BV2 microglia. It was also shown that HDAC inhibitor (HDACi) treatment mimics the effects of Runx1t1 knockdown on microglial proliferation, confirming that microglial proliferation is associated with Runx1t1 expression and HDACs activity. Further, Runx1t1 and HDACs were shown to promote neurotoxic effect of microglia by repressing expression of LAT2, L-aminoacid transporter-2 (cationic amino acid transporter, y+ system), which normally inhibits NO production. This was confirmed by chromatin immunoprecipitation (ChIP) assay, which revealed that Runx1t1 binds to the promoter region of LAT2 and this binding increased upon microglial activation. However, the enhanced binding of Runx1t1 to the LAT2 promoter could not repress the LAT2 expression when the BV2 microglia cells were treated with HDACi, indicating that Runx1t1 requires HDACs to transcriptionally repress the expression of LAT2.In conclusion, it is suggested that Runx1t1 controls proliferation and the neurotoxic effect of microglia by epigenetically regulating Cdk4 and LAT2 via its interaction with HDACs. | | 24586690
|
Flexibilide obtained from cultured soft coral has anti-neuroinflammatory and analgesic effects through the upregulation of spinal transforming growth factor-β1 in neuropathic rats.
Chen, NF; Huang, SY; Lu, CH; Chen, CL; Feng, CW; Chen, CH; Hung, HC; Lin, YY; Sung, PJ; Sung, CS; Yang, SN; Wang, HM; Chang, YC; Sheu, JH; Chen, WF; Wen, ZH
Marine drugs
12
3792-817
2014
Show Abstract
Chronic neuroinflammation plays an important role in the development and maintenance of neuropathic pain. The compound flexibilide, which can be obtained from cultured soft coral, possesses anti-inflammatory and analgesic effects in the rat carrageenan peripheral inflammation model. In the present study, we investigated the antinociceptive properties of flexibilide in the rat chronic constriction injury (CCI) model of neuropathic pain. First, we found that a single intrathecal (i.t.) administration of flexibilide significantly attenuated CCI-induced thermal hyperalgesia at 14 days after surgery. Second, i.t. administration of 10-μg flexibilide twice daily was able to prevent the development of thermal hyperalgesia and weight-bearing deficits in CCI rats. Third, i.t. flexibilide significantly inhibited CCI-induced activation of microglia and astrocytes, as well as the upregulated proinflammatory enzyme, inducible nitric oxide synthase, in the ipsilateral spinal dorsal horn. Furthermore, flexibilide attenuated the CCI-induced downregulation of spinal transforming growth factor-β1 (TGF-β1) at 14 days after surgery. Finally, i.t. SB431542, a selective inhibitor of TGF-β type I receptor, blocked the analgesic effects of flexibilide in CCI rats. Our results suggest that flexibilide may serve as a therapeutic agent for neuropathic pain. In addition, spinal TGF-β1 may be involved in the anti-neuroinflammatory and analgesic effects of flexibilide. | | 24979268
|
Adenosine kinase, glutamine synthetase and EAAT2 as gene therapy targets for temporal lobe epilepsy.
Young, D; Fong, DM; Lawlor, PA; Wu, A; Mouravlev, A; McRae, M; Glass, M; Dragunow, M; During, MJ
Gene therapy
21
1029-40
2014
Show Abstract
Astrocytes are an attractive cell target for gene therapy, but the validation of new therapeutic candidates is needed. We determined whether adeno-associated viral (AAV) vector-mediated overexpression of glutamine synthetase (GS) or excitatory amino-acid transporter 2 (EAAT2), or expression of microRNA targeting adenosine kinase (miR-ADK) in hippocampal astrocytes in the rat brain could modulate susceptibility to kainate-induced seizures and neuronal cell loss. Transgene expression was found predominantly in astrocytes following direct injection of glial-targeting AAV9 vectors by 3 weeks postinjection. ADK expression in miR-ADK vector-injected rats was reduced by 94-96% and was associated with an ~50% reduction in the duration of kainate-induced seizures and greater protection of dentate hilar neurons but not CA3 neurons compared with miR-control vector-injected rats. In contrast, infusion of AAV-GS and EAAT2 vectors did not afford any protection against seizures or neuronal damage as the level of transcriptional activity of the glial fibrillary acidic promoter was too low to drive any significant increase in transgenic GS or EAAT2 relative to the high endogenous levels of these proteins. Our findings support ADK as a prime therapeutic target for gene therapy of temporal lobe epilepsy and suggest that alternative approaches including the use of stronger glial promoters are needed to increase transgenic GS and EAAT2 expression to levels that may be required to affect seizure induction and propagation. | | 25231174
|
Toll-like receptor 4 mediates microglial activation and production of inflammatory mediators in neonatal rat brain following hypoxia: role of TLR4 in hypoxic microglia.
Yao, L; Kan, EM; Lu, J; Hao, A; Dheen, ST; Kaur, C; Ling, EA
Journal of neuroinflammation
10
23
2013
Show Abstract
Hypoxia induces microglial activation which causes damage to the developing brain. Microglia derived inflammatory mediators may contribute to this process. Toll-like receptor 4 (TLR4) has been reported to induce microglial activation and cytokines production in brain injuries; however, its role in hypoxic injury remains uncertain. We investigate here TLR4 expression and its roles in neuroinflammation in neonatal rats following hypoxic injury.One day old Wistar rats were subjected to hypoxia for 2 h. Primary cultured microglia and BV-2 cells were subjected to hypoxia for different durations. TLR4 expression in microglia was determined by RT-PCR, western blot and immunofluorescence staining. Small interfering RNA (siRNA) transfection and antibody neutralization were employed to downregulate TLR4 in BV-2 and primary culture. mRNA and protein expression of tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β) and inducible nitric oxide synthase (iNOS) was assessed. Reactive oxygen species (ROS), nitric oxide (NO) and NF-κB levels were determined by flow cytometry, colorimetric and ELISA assays respectively. Hypoxia-inducible factor-1 alpha (HIF-1α) mRNA and protein expression was quantified and where necessary, the protein expression was depleted by antibody neutralization. In vivo inhibition of TLR4 with CLI-095 injection was carried out followed by investigation of inflammatory mediators expression via double immunofluorescence staining.TLR4 immunofluorescence and protein expression in the corpus callosum and cerebellum in neonatal microglia were markedly enhanced post-hypoxia. In vitro, TLR4 protein expression was significantly increased in both primary microglia and BV-2 cells post-hypoxia. TLR4 neutralization in primary cultured microglia attenuated the hypoxia-induced expression of TNF-α, IL-1β and iNOS. siRNA knockdown of TLR4 reduced hypoxia-induced upregulation of TNF-α, IL-1β, iNOS, ROS and NO in BV-2 cells. TLR4 downregulation-mediated inhibition of inflammatory cytokines in primary microglia and BV-2 cells was accompanied by the suppression of NF-κB activation. Furthermore, HIF-1α antibody neutralization attenuated the increase of TLR4 expression in hypoxic BV-2 cells. TLR4 inhibition in vivo attenuated the immunoexpression of TNF-α, IL-1β and iNOS on microglia post-hypoxia.Activated microglia TLR4 expression mediated neuroinflammation via a NF-κB signaling pathway in response to hypoxia. Hence, microglia TLR4 presents as a potential therapeutic target for neonatal hypoxia brain injuries. | Immunohistochemistry | 23388509
|
PPARγ activation blocks development and reduces established neuropathic pain in rats.
Morgenweck, J; Griggs, RB; Donahue, RR; Zadina, JE; Taylor, BK
Neuropharmacology
70
236-46
2013
Show Abstract
Peroxisome proliferator-activated receptor gamma (PPARγ) is emerging as a new pharmacotherapeutic target for chronic pain. When oral (3-30 mg/kg/day in chow for 7 wk) or twice-daily intraperitoneal (1-10 mg/kg/day for 2 wk) administration began before spared nerve injury (SNI), pioglitazone, a PPARγ agonist, dose-dependently prevented multiple behavioral signs of somatosensory hypersensitivity. The highest dose of intraperitoneal pioglitazone did not produce ataxia or reductions in transient mechanical and heat nociception, indicating that inhibitory effects on hypersensitivity were not secondary to adverse drug-induced behaviors or antinociception. Inhibitory effects on hypersensitivity persisted at least one week beyond cessation of pioglitazone administration, suggestive of long-lasting effects on gene expression. Blockade of PPARγ with GW9662, an irreversible and selective PPARγ antagonist, dose-dependently reduced the inhibitory effect of pioglitazone on hypersensitivity, indicating a PPARγ-dependent action. Remarkably, a single preemptive injection of pioglitazone 15 min before SNI attenuated hypersensitivity for at least 2 weeks; this was enhanced with a second injection delivered 12 h after SNI. Pioglitazone injections beginning after SNI also reduced hypersensitivity, albeit to a lesser degree than preemptive treatment. Intraperitoneal pioglitazone significantly reduced the nerve injury-induced up-regulation of cd11b, GFAP, and p-p38 in the dorsal horn, indicating a mechanism of action involving spinal microglia and/or astrocyte activation. Oral pioglitazone significantly reduced touch stimulus-evoked phospho-extracellular signal-related kinase (p-ERK) in lamina I-II, indicating a mechanism of action involving inhibition of central sensitization. We conclude that pioglitazone reduces spinal glial and stimulus-evoked p-ERK activation and that PPARγ activation blocks the development of and reduces established neuropathic pain. | | 23415633
|