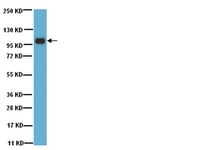
Cezanne regulates E2F1-dependent HIF2α expression.
Moniz, S; Bandarra, D; Biddlestone, J; Campbell, KJ; Komander, D; Bremm, A; Rocha, S
Journal of cell science
128
3082-93
2015
Show Abstract
Mechanisms regulating protein degradation ensure the correct and timely expression of transcription factors such as hypoxia inducible factor (HIF). Under normal O2 tension, HIFα subunits are targeted for proteasomal degradation, mainly through vHL-dependent ubiquitylation. Deubiquitylases are responsible for reversing this process. Although the mechanism and regulation of HIFα by ubiquitin-dependent proteasomal degradation has been the object of many studies, little is known about the role of deubiquitylases. Here, we show that expression of HIF2α (encoded by EPAS1) is regulated by the deubiquitylase Cezanne (also known as OTUD7B) in an E2F1-dependent manner. Knockdown of Cezanne downregulates HIF2α mRNA, protein and activity independently of hypoxia and proteasomal degradation. Mechanistically, expression of the HIF2α gene is controlled directly by E2F1, and Cezanne regulates the stability of E2F1. Exogenous E2F1 can rescue HIF2α transcript and protein expression when Cezanne is depleted. Taken together, these data reveal a novel mechanism for the regulation of the expression of HIF2α, demonstrating that the HIF2α promoter is regulated by E2F1 directly and that Cezanne regulates HIF2α expression through control of E2F1 levels. Our results thus suggest that HIF2α is controlled transcriptionally in a cell-cycle-dependent manner and in response to oncogenic signalling. | | 26148512
 |
An Sp1 Modulated Regulatory Region Unique to Higher Primates Regulates Human Androgen Receptor Promoter Activity in Prostate Cancer Cells.
Hay, CW; Hunter, I; MacKenzie, A; McEwan, IJ
PloS one
10
e0139990
2015
Show Abstract
Androgen receptor (AR) mediated signalling is necessary for normal development of the prostate gland and also drives prostate cancer (PCa) cell growth and survival, with many studies showing a correlation between increased receptor levels and therapy resistance with progression to fatal castrate recurrent PCa (CRPC). Although it has been held for some time that the transcription factor Sp1 is the main stimulator of AR gene transcription, comprehensive knowledge of the regulation of the AR gene remains incomplete. Here we describe and characterise in detail two novel active regulatory elements in the 5'UTR of the human AR gene. Both of these elements contain overlapping binding sites for the positive transcription factor Sp1 and the repressor protein pur-α. Aberrant cell signalling is characteristic of PCa and the transcriptional activity of the AR promoter in PCa cells is dependent upon the relative amounts of the two transcription factors. Together with our corroboration of the dominant role of Sp1, the findings support the rationale of targeting this transcription factor to inhibit tumour progression. This should be of particular therapeutic relevance in CRPC where the levels of the repressor pur-α are reduced. | | 26448047
|
CR6-interacting factor 1 is a key regulator in Aβ-induced mitochondrial disruption and pathogenesis of Alzheimer's disease.
Byun, J; Son, SM; Cha, MY; Shong, M; Hwang, YJ; Kim, Y; Ryu, H; Moon, M; Kim, KS; Mook-Jung, I
Cell death and differentiation
22
959-73
2015
Show Abstract
Mitochondrial dysfunction, often characterized by massive fission and other morphological abnormalities, is a well-known risk factor for Alzheimer's disease (AD). One causative mechanism underlying AD-associated mitochondrial dysfunction is thought to be amyloid-β (Aβ), yet the pathways between Aβ and mitochondrial dysfunction remain elusive. In this study, we report that CR6-interacting factor 1 (Crif1), a mitochondrial inner membrane protein, is a key player in Aβ-induced mitochondrial dysfunction. Specifically, we found that Crif1 levels were downregulated in the pathological regions of Tg6799 mice brains, wherein overexpressed Aβ undergoes self-aggregation. Downregulation of Crif1 was similarly observed in human AD brains as well as in SH-SY5Y cells treated with Aβ. In addition, knockdown of Crif1, using RNA interference, induced mitochondrial dysfunction with phenotypes similar to those observed in Aβ-treated cells. Conversely, Crif1 overexpression prevented Aβ-induced mitochondrial dysfunction and cell death. Finally, we show that Aβ-induced downregulation of Crif1 is mediated by enhanced reactive oxygen species (ROS) and ROS-dependent sumoylation of the transcription factor specificity protein 1 (Sp1). These results identify the ROS-Sp1-Crif1 pathway to be a new mechanism underlying Aβ-induced mitochondrial dysfunction and suggest that ROS-mediated downregulation of Crif1 is a crucial event in AD pathology. We propose that Crif1 may serve as a novel therapeutic target in the treatment of AD. | | 25361083
|
Erythroid induction of K562 cells treated with mithramycin is associated with inhibition of raptor gene transcription and mammalian target of rapamycin complex 1 (mTORC1) functions.
Finotti, A; Bianchi, N; Fabbri, E; Borgatti, M; Breveglieri, G; Gasparello, J; Gambari, R
Pharmacological research
91
57-68
2015
Show Abstract
Rapamycin, an inhibitor of mTOR activity, is a potent inducer of erythroid differentiation and fetal hemoglobin production in β-thalassemic patients. Mithramycin (MTH) was studied to see if this inducer of K562 differentiation also operates through inhibition of mTOR. We can conclude from the study that the mTOR pathway is among the major transcript classes affected by mithramycin-treatment in K562 cells and a sharp decrease of raptor protein production and p70S6 kinase is detectable in mithramycin treated K562 cells. The promoter sequence of the raptor gene contains several Sp1 binding sites which may explain its mechanism of action. We hypothesize that the G+C-selective DNA-binding drug mithramycin is able to interact with these sequences and to inhibit the binding of Sp1 to the raptor promoter due to the following results: (a) MTH strongly inhibits the interactions between Sp1 and Sp1-binding sites of the raptor promoter (studied by electrophoretic mobility shift assays, EMSA); (b) MTH strongly reduces the recruitment of Sp1 transcription factor to the raptor promoter in intact K562 cells (studied by chromatin immunoprecipitation experiments, ChIP); (c) Sp1 decoy oligonucleotides are able to specifically inhibit raptor mRNA accumulation in K562 cells. In conclusion, raptor gene expression is involved in mithramycin-mediated induction of erythroid differentiation of K562 cells and one of its mechanism of action is the inhibition of Sp1 binding to the raptor promoter. | | 25478892
|
Down-regulation of Sp1 suppresses cell proliferation, clonogenicity and the expressions of stem cell markers in nasopharyngeal carcinoma.
Zhang, JP; Zhang, H; Wang, HB; Li, YX; Liu, GH; Xing, S; Li, MZ; Zeng, MS
Journal of translational medicine
12
222
2014
Show Abstract
Transcription factor Sp1 is multifaceted, with the ability to function as an oncogene or a tumor suppressor, depending on the cellular context. We previously reported that Sp1 is required for the transcriptional activation of the key oncogenes in nasopharyngeal carcinoma (NPC), including B-lymphoma mouse Moloney leukemia virus insertion region 1 (Bmi1) and centromere protein H (CENPH), but the role of Sp1 and its underlying mechanisms in NPC remained largely unexplored. The objective of this study was to investigate the cellular function of Sp1 and to verify the clinical significance of Sp1 as a potential therapeutic target in NPC.The levels of Sp1 in the normal primary nasopharyngeal epithelial cells (NPECs) and NPC cell lines were analyzed by Quantitative Real-time RT-PCR (qRT-PCR) and Western blot. The location and expression of Sp1 in the NPC tissues were detected by immunohistochemistry staining (IHC). The effect of Sp1 knockdown on the cell proliferation, clonogenicity, anchorage-independent growth and the stem-cell like phenotype in NPC cells were evaluated by MTT, flow cytometry, clonogenicity analysis and sphere formation assay.The mRNA and protein levels of Sp1 were elevated in NPC cell lines than in the normal primary NPECs. Higher expression of Sp1 was found in NPC tissues with advanced clinical stage (P=0.00036). Either inhibition of Sp1 activity by mithramycin A, the FDA-approved chemotherapeutic anticancer drug or Sp1 silencing by two distinct siRNA against Sp1 suppressed the growth of NPC cells. Mechanism analysis revealed that Sp1 silencing may suppress cell proliferation, clonogenicity, anchorage-independent growth and the stem-cell like phenotype through inducing the expression of p27 and p21, and impairing the expressions of the critical stem cell transcription factors (SCTFs), including Bmi1, c-Myc and KLF4 in NPC cells.Sp1 was enriched in advanced NPC tissues and silencing of Sp1 significantly inhibited cell proliferation, clonogenicity, anchorage-independent growth and the stem-cell like phenotype of NPC cells, suggesting Sp1 may serve as an appealing drug target for NPC. | | 25099028
|
Regulatory mechanism of endothelin receptor B in the cerebral arteries after focal cerebral ischemia.
Grell, AS; Thigarajah, R; Edvinsson, L; Samraj, AK
PloS one
9
e113624
2014
Show Abstract
Increased expression of endothelin receptor type B (ETBR), a vasoactive receptor, has recently been implied in the reduced cerebral blood flow and exacerbated neuronal damage after ischemia-reperfusion (I/R). The study explores the regulatory mechanisms of ETBR to identify drug targets to restore normal cerebral artery contractile function as part of successful neuroprotective therapy.We have employed in vitro methods on human and rat cerebral arteries to study the regulatory mechanisms and the efficacy of target selective inhibitor, Mithramycin A (MitA), to block the ETBR mediated contractile properties. Later, middle cerebral artery occluded (MCAO) rats were used to substantiate the observations. Quantative PCR, immunohistochemistry, western blot and wire myograph methods were employed to study the expression and contractile properties of cerebral arteries.Increased expression of specificity protein (Sp1) was observed in human and rat cerebral arteries after organ culture, strongly correlating with the ETBR upregulation. Similar observations were made in MCAO rats. Treatment with MitA, a Sp1 specific inhibitor, significantly downregulated the ETBR mRNA and protein levels. It also significantly reduced the ETBR mediated cerebrovascular contractility. Detailed analysis indicated that ERK1/2 mediated phosphorylation of Sp1 might be essential for ETBR transcription.Transcription factor Sp1 regulates the ETBR mediated vasoconstriction in focal cerebral ischemia via MEK-ERK signaling, which is also conserved in humans. The results show that MitA can effectively be used to block ETBR mediated vasoconstriction as a supplement to an existing ischemic stroke therapy. | Immunohistochemistry | 25479176
|
Contribution of transcription factor, SP1, to the promotion of HB-EGF expression in defense mechanism against the treatment of irinotecan in ovarian clear cell carcinoma.
Miyata, K; Yotsumoto, F; Nam, SO; Odawara, T; Manabe, S; Ishikawa, T; Itamochi, H; Kigawa, J; Takada, S; Asahara, H; Kuroki, M; Miyamoto, S
Cancer medicine
3
1159-69
2014
Show Abstract
Ovarian clear cell carcinoma (OCCC) is a worst histological subtype than other ovarian malignant tumor. Heparin-binding epidermal growth factor-like growth factor (HB-EGF) is a promising target for ovarian cancer therapy. The aims of this study were to validate the efficacy of HB-EGF-targeted therapy for OCCC and to identify the transcription factor that contributed to the induction of HB-EGF by SN38 treatment in OCCC cells. HB-EGF was highly expressed in OCCC cells, and an increase of HB-EGF was induced by SN38 which had only antitumor effect among conventional anticancer agents on OCCC. A specific inhibitor of HB-EGF, a cross-reacting material 197 (CRM197), led to a synergistic increase in the number of apoptotic OCCC cells with the treatment of SN38. The luciferase assay with 5'-deletion promoter constructs identified a GC-rich element between -125 and -178 (the distal transcription start site was denoted +1) as a cis-regulatory region, and the treatment of SN38 induced luciferase activity in this region. An in silico and chromatin immunoprecipitation analysis estimated that SP1 bound to the cis-regulatory region of HB-EGF in OCCC cells. Real-time PCR and cell viability assays showed that the transfection of a small interfering RNA targeting SP1 suppressed the expression of HB-EGF induced by SN38, resulting in the enhanced sensitivity of SN38. Taken together, these results indicate that induction of HB-EGF expression contributed to defense mechanism against treatment of SN38 through the transcriptional activity of SP1 in OCCC cells. | Western Blotting | 25060396
|
HDAC1 and Klf4 interplay critically regulates human myeloid leukemia cell proliferation.
Huang, Y; Chen, J; Lu, C; Han, J; Wang, G; Song, C; Zhu, S; Wang, C; Li, G; Kang, J; Wang, J
Cell death & disease
5
e1491
2014
Show Abstract
Acute myeloid leukemia (AML) is recognized as a complex disease of hematopoietic stem cell disorders, but its pathogenesis mechanisms, diagnosis, and treatment remain unclear. General histone deacetylase (HDAC) inhibitors have been used in blood cancers including AML, but the lack of gene specificity greatly limits their anti-cancer effects and clinical applications. Here, we found that HDAC1 expression was negatively correlated with that of Krüppel-like factor 4 (Klf4) and that AML patients with lower HDAC1 level had better prognosis. Further, knockdown of HDAC1 in leukemia cells K562, HL-60, and U937 significantly increased Klf4 expression and inhibited cell cycle progression and cell proliferation, similar results were found for HDAC inhibitors (VPA and mocetinostat). Moreover, overexpression or knockdown of Klf4 could markedly block the effects of HDAC1 overexpression or knockdown on leukemia cells in vitro and in vivo, respectively. Mechanistic analyses demonstrated that HDAC1 and Klf4 competitively bound to the promoter region of Klf4 and oppositely regulated Klf4 expression in myeloid leukemia. We identified HDAC1 as a potential specific target for repressing cell proliferation and inducing cell cycle arrest through interplay and modulation of Klf4 expression, suggests that HDAC1 and Klf4 are potential new molecular markers and targets for clinical diagnosis, prognosis, and treatment of myeloid leukemia. | | 25341045
|
Egr-1 regulates the transcription of NGX6 gene through a Sp1/Egr-1 overlapping site in the promoter.
Liu, M; Wang, X; Peng, Y; Shen, S; Li, G
BMC molecular biology
15
14
2014
Show Abstract
As a novel candidate metastasis suppressor gene, Nasopharyngeal carcinoma-associated gene 6 (NGX6) is involved in cellular growth, cell cycle progression and tumor angiogenesis. Previous studies have shown that NGX6 gene is down-regulated in colorectal cancer (CRC). However, little is known about its transcriptional regulation.We defined the minimal promoter of NGX6 gene in a 186-bp region (from-86 to +100) through mutation construct methods and luciferase assays. Results from Electrophoretic mobility shift assays (EMSA) and Chromatin immunoprecipitation (ChIP) revealed that Early growth response gene 1 (Egr-1) binds to the Sp1/Egr-1 overlapping site of NGX6 minimal promoter. Overexpression of Egr-1 increased the promoter activity and mRNA level of NGX6 gene; while knock-down of endogenous Egr-1 decreased mRNA expression of NGX6 gene.These results demonstrate that Egr-1 regulates NGX6 gene transcription through an overlapping Sp1/Egr-1 binding site as a positive regulator of NGX6 gene. | | 25029911
|
Loss of Jak2 Impairs Endothelial Function by Attenuating Raf-1/MEK1/Sp-1 Signaling Along with Altered eNOS Activities.
Yang, Ping, et al.
Am. J. Pathol., 183: 617-25 (2013)
2013
Show Abstract
A number of inhibitors have been used to dissect the functional relevance of Jak2 in endothelial homeostasis, with disparate results. Given that Jak2 deficiency leads to embryonic lethality, the exact role of Jak2 in the regulation of postnatal endothelial function is yet to be fully elucidated. We generated a model in which Jak2 deficiency can be induced by tamoxifen in adult mice. Loss of Jak2 significantly impaired endothelium-dependent response capacity for vasodilators. Matrigel plug assays indicated a notable decrease in endothelial angiogenic function in Jak2-deficient mice. Studies in a hindlimb ischemic model indicated that Jak2 activity is likely to be a prerequisite for prompt perfusion recovery, based on the concordance of temporal changes in Jak2 expression during the course of ischemic injury and perfusion recovery. A remarkable delay in perfusion recovery, along with reduced capillary and arteriole formation, was observed in Jak2-deficient mice. Antibody array studies indicated that loss of Jak2 led to repressed eNOS expression. In mechanistic studies, Jak2 deficiency attenuated Raf-1/MEK1 signaling, which then reduced activity of Sp-1, an essential transcription factor responsible for eNOS expression. These data are important not only for understanding the exact role that Jak2 plays in endothelial homeostasis, but also for assessing Jak2-based therapeutic strategies in a variety of clinical settings. | | 23747947
|