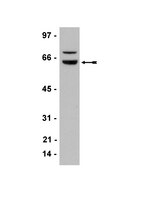
Wilms' tumor gene 1 (WT1) silencing inhibits proliferation of malignant peripheral nerve sheath tumor sNF96.2 cell line.
Parenti, R; Cardile, V; Graziano, AC; Parenti, C; Venuti, A; Bertuccio, MP; Furno, DL; Magro, G
PloS one
9
e114333
2014
Show Abstract
Wilms' tumor gene 1 (WT1) plays complex roles in tumorigenesis, acting as tumor suppressor gene or an oncogene depending on the cellular context. WT1 expression has been variably reported in both benign and malignant peripheral nerve sheath tumors (MPNSTs) by means of immunohistochemistry. The aim of the present study was to characterize its potential pathogenetic role in these relatively uncommon malignant tumors. Firstly, immunohistochemical analyses in MPNST sNF96.2 cell line showed strong WT1 staining in nuclear and perinuclear areas of neoplastic cells. Thus, we investigated the effects of silencing WT1 by RNA interference. Through Western Blot analysis and proliferation assay we found that WT1 knockdown leads to the reduction of cell growth in a time- and dose-dependent manner. siWT1 inhibited proliferation of sNF96.2 cell lines likely by influencing cell cycle progression through a decrease in the protein levels of cyclin D1 and inhibition of Akt phosphorylation compared to the control cells. These results indicate that WT1 knockdown attenuates the biological behavior of MPNST cells by decreasing Akt activity, demonstrating that WT1 is involved in the development and progression of MPNSTs. Thus, WT1 is suggested to serve as a potential therapeutic target for MPNSTs. | Immunofluorescence | 25474318
 |
DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia.
Rampal, R; Alkalin, A; Madzo, J; Vasanthakumar, A; Pronier, E; Patel, J; Li, Y; Ahn, J; Abdel-Wahab, O; Shih, A; Lu, C; Ward, PS; Tsai, JJ; Hricik, T; Tosello, V; Tallman, JE; Zhao, X; Daniels, D; Dai, Q; Ciminio, L; Aifantis, I; He, C; Fuks, F; Tallman, MS; Ferrando, A; Nimer, S; Paietta, E; Thompson, CB; Licht, JD; Mason, CE; Godley, LA; Melnick, A; Figueroa, ME; Levine, RL
Cell reports
9
1841-55
2014
Show Abstract
Somatic mutations in IDH1/IDH2 and TET2 result in impaired TET2-mediated conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC). The observation that WT1 inactivating mutations anticorrelate with TET2/IDH1/IDH2 mutations in acute myeloid leukemia (AML) led us to hypothesize that WT1 mutations may impact TET2 function. WT1 mutant AML patients have reduced 5hmC levels similar to TET2/IDH1/IDH2 mutant AML. These mutations are characterized by convergent, site-specific alterations in DNA hydroxymethylation, which drive differential gene expression more than alterations in DNA promoter methylation. WT1 overexpression increases global levels of 5hmC, and WT1 silencing reduced 5hmC levels. WT1 physically interacts with TET2 and TET3, and WT1 loss of function results in a similar hematopoietic differentiation phenotype as observed with TET2 deficiency. These data provide a role for WT1 in regulating DNA hydroxymethylation and suggest that TET2 IDH1/IDH2 and WT1 mutations define an AML subtype defined by dysregulated DNA hydroxymethylation. | | 25482556
|
De novo lumen formation and elongation in the developing nephron: a central role for afadin in apical polarity.
Yang, Z; Zimmerman, S; Brakeman, PR; Beaudoin, GM; Reichardt, LF; Marciano, DK
Development (Cambridge, England)
140
1774-84
2013
Show Abstract
A fundamental process in biology is the de novo formation and morphogenesis of polarized tubules. Although these processes are essential for the formation of multiple metazoan organ systems, little is known about the molecular mechanisms that regulate them. In this study, we have characterized several steps in tubule formation and morphogenesis using the mouse kidney as a model system. We report that kidney mesenchymal cells contain discrete Par3-expressing membrane microdomains that become restricted to an apical domain, coinciding with lumen formation. Once lumen formation has been initiated, elongation occurs by simultaneous extension and additional de novo lumen generation. We demonstrate that lumen formation and elongation require afadin, a nectin adaptor protein implicated in adherens junction formation. Mice that lack afadin in nephron precursors show evidence of Par3-expressing membrane microdomains, but fail to develop normal apical-basal polarity and generate a continuous lumen. Absence of afadin led to delayed and diminished integration of nectin complexes and failure to recruit R-cadherin. Furthermore, we demonstrate that afadin is required for Par complex formation. Together, these results suggest that afadin acts upstream of the Par complex to regulate the integration and/or coalescence of membrane microdomains, thereby establishing apical-basal polarity and lumen formation/elongation during kidney tubulogenesis. | | 23487309
|
aPKCλ/ι and aPKCζ contribute to podocyte differentiation and glomerular maturation.
Hartleben, B; Widmeier, E; Suhm, M; Worthmann, K; Schell, C; Helmstädter, M; Wiech, T; Walz, G; Leitges, M; Schiffer, M; Huber, TB
Journal of the American Society of Nephrology : JASN
24
253-67
2013
Show Abstract
Precise positioning of the highly complex interdigitating podocyte foot processes is critical to form the normal glomerular filtration barrier, but the molecular programs driving this process are unknown. The protein atypical protein kinase C (aPKC)--a component of the Par complex, which localizes to tight junctions and interacts with slit diaphragm proteins--may play a role. Here, we found that the combined deletion of the aPKCλ/ι and aPKCζ isoforms in podocytes associated with incorrectly positioned centrosomes and Golgi apparatus and mislocalized molecules of the slit diaphragm. Furthermore, aPKC-deficient podocytes failed to form the normal network of foot processes, leading to defective glomerular maturation with incomplete capillary formation and mesangiolysis. Our results suggest that aPKC isoforms orchestrate the formation of the podocyte processes essential for normal glomerular development and kidney function. Defective aPKC signaling results in a dramatically simplified glomerular architecture, causing severe proteinuria and perinatal death. | | 23334392
|
Loss of podocyte aPKClambda/iota causes polarity defects and nephrotic syndrome.
Tobias B Huber,Björn Hartleben,Kirstin Winkelmann,Lisa Schneider,Jan U Becker,Michael Leitges,Gerd Walz,Hermann Haller,Mario Schiffer,Björn Hartleben
Journal of the American Society of Nephrology : JASN
20
2009
Show Abstract
Atypical protein kinase C (aPKC) is a central component of the evolutionarily conserved Par3-Par6-aPKC complex, one of the fundamental regulators of cell polarity. We recently demonstrated that these proteins interact with Neph-nephrin molecules at the slit diaphragm of the glomerular filtration barrier. Here, we report that podocyte-specific deletion of aPKClambda/iota in mice results in severe proteinuria, nephrotic syndrome, and death at 4 to 5 wk after birth. Podocyte foot processes of knockout mice developed structural defects, including mislocalization of the slit diaphragm. In the glomerulus, aPKClambda/iota was primarily expressed in developing glomerular epithelial cells and podocyte foot processes. Interestingly, under physiologic conditions, aPKClambda/iota translocated from the apical surface to the basolateral side of developing podocytes, and this translocation preceded the development of foot processes and formation of slit diaphragms. Supporting a critical role for aPKClambda/iota in the maintenance of slit diaphragms and podocyte foot processes, aPKClambda/iota associated with the Neph-nephrin slit diaphragm complex and localized to the tips of filopodia and leading edges of cultured podocytes. These results suggest that aPKC signaling is fundamental to glomerular maintenance and development. Full Text Article | | 19279126
|