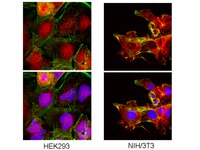
Acid ceramidase (ASAH1) is a global regulator of steroidogenic capacity and adrenocortical gene expression.
Lucki, NC; Bandyopadhyay, S; Wang, E; Merrill, AH; Sewer, MB
Mol Endocrinol
26
228-43
2012
Show Abstract
In H295R human adrenocortical cells, ACTH rapidly activates ceramide (Cer) and sphingosine (SPH) turnover with a concomitant increase in SPH-1-phosphate secretion. These bioactive lipids modulate adrenocortical steroidogenesis, primarily by acting as second messengers in the protein kinase A/cAMP-dependent pathway. Acid ceramidase (ASAH1) directly regulates the intracellular balance of Cer, SPH, and SPH-1-phosphate by catalyzing the hydrolysis of Cer into SPH. ACTH/cAMP signaling stimulates ASAH1 transcription and activity, supporting a role for this enzyme in glucocorticoid production. Here, the role of ASAH1 in regulating steroidogenic capacity was examined using a tetracycline-inducible ASAH1 short hairpin RNA H295R human adrenocortical stable cell line. We show that ASAH1 suppression increases the transcription of multiple steroidogenic genes, including Cytochrome P450 monooxygenase (CYP)17A1, CYP11B1/2, CYP21A2, steroidogenic acute regulatory protein, hormone-sensitive lipase, 18-kDa translocator protein, and the melanocortin-2 receptor. Induced gene expression positively correlated with enhanced histone H3 acetylation at target promoters. Repression of ASAH1 expression also induced the expression of members of the nuclear receptor nuclear receptor subfamily 4 (NR4A) family while concomitantly suppressing the expression of dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1. ASAH1 knockdown altered the expression of genes involved in sphingolipid metabolism and changed the cellular amounts of distinct sphingolipid species. Finally, ASAH1 silencing increased basal and cAMP-dependent cortisol and dehydroepiandrosterone secretion, establishing ASAH1 as a pivotal regulator of steroidogenic capacity in the human adrenal cortex. | | 22261821
 |
Inhibition of GSK3β improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome.
Guo, W; Murthy, AC; Zhang, L; Johnson, EB; Schaller, EG; Allan, AM; Zhao, X
Human molecular genetics
21
681-91
2012
Show Abstract
Fragile X syndrome (FXS), a common inherited form of intellectual disability with learning deficits, results from a loss of fragile X mental retardation protein (FMRP). Despite extensive research, treatment options for FXS remain limited. Since FMRP is known to play an important role in adult hippocampal neurogenesis and hippocampus-dependent learning and FMRP regulates the adult neural stem cell fate through the translational regulation of glycogen synthase kinase 3β (GSK3β), we investigated the effects of a GSK3β inhibitor, SB216763, on Fmr1 knockout mice (Fmr1 KO). We found that the inhibition of GSK3β could reverse the hippocampus-dependent learning deficits and rescue adult hippocampal neurogenesis at multiple stages in Fmr1 KO mice. Our results point to GSK3β inhibition as a potential treatment for the learning deficits seen in FXS. | Western Blotting | 22048960
|