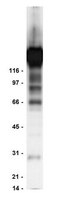
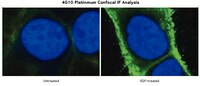
Phosphorylation site dynamics of early T-cell receptor signaling.
Chylek, LA; Akimov, V; Dengjel, J; Rigbolt, KT; Hu, B; Hlavacek, WS; Blagoev, B
PloS one
9
e104240
2014
Show Abstract
In adaptive immune responses, T-cell receptor (TCR) signaling impacts multiple cellular processes and results in T-cell differentiation, proliferation, and cytokine production. Although individual protein-protein interactions and phosphorylation events have been studied extensively, we lack a systems-level understanding of how these components cooperate to control signaling dynamics, especially during the crucial first seconds of stimulation. Here, we used quantitative proteomics to characterize reshaping of the T-cell phosphoproteome in response to TCR/CD28 co-stimulation, and found that diverse dynamic patterns emerge within seconds. We detected phosphorylation dynamics as early as 5 s and observed widespread regulation of key TCR signaling proteins by 30 s. Development of a computational model pointed to the presence of novel regulatory mechanisms controlling phosphorylation of sites with central roles in TCR signaling. The model was used to generate predictions suggesting unexpected roles for the phosphatase PTPN6 (SHP-1) and shortcut recruitment of the actin regulator WAS. Predictions were validated experimentally. This integration of proteomics and modeling illustrates a novel, generalizable framework for solidifying quantitative understanding of a signaling network and for elucidating missing links. | Immunoprecipitation | 25147952
 |
Identification of fibroblast growth factor receptor 3 (FGFR3) as a protein receptor for botulinum neurotoxin serotype A (BoNT/A).
Jacky, BP; Garay, PE; Dupuy, J; Nelson, JB; Cai, B; Molina, Y; Wang, J; Steward, LE; Broide, RS; Francis, J; Aoki, KR; Stevens, RC; Fernández-Salas, E
PLoS pathogens
9
e1003369
2013
Show Abstract
Botulinum neurotoxin serotype A (BoNT/A) causes transient muscle paralysis by entering motor nerve terminals (MNTs) where it cleaves the SNARE protein Synaptosomal-associated protein 25 (SNAP25206) to yield SNAP25197. Cleavage of SNAP25 results in blockage of synaptic vesicle fusion and inhibition of the release of acetylcholine. The specific uptake of BoNT/A into pre-synaptic nerve terminals is a tightly controlled multistep process, involving a combination of high and low affinity receptors. Interestingly, the C-terminal binding domain region of BoNT/A, HC/A, is homologous to fibroblast growth factors (FGFs), making it a possible ligand for Fibroblast Growth Factor Receptors (FGFRs). Here we present data supporting the identification of Fibroblast Growth Factor Receptor 3 (FGFR3) as a high affinity receptor for BoNT/A in neuronal cells. HC/A binds with high affinity to the two extra-cellular loops of FGFR3 and acts similar to an agonist ligand for FGFR3, resulting in phosphorylation of the receptor. Native ligands for FGFR3; FGF1, FGF2, and FGF9 compete for binding to FGFR3 and block BoNT/A cellular uptake. These findings show that FGFR3 plays a pivotal role in the specific uptake of BoNT/A across the cell membrane being part of a larger receptor complex involving ganglioside- and protein-protein interactions. | | 23696738
|
The neuroblastoma ALK(I1250T) mutation is a kinase-dead RTK in vitro and in vivo.
Schönherr, C; Ruuth, K; Eriksson, T; Yamazaki, Y; Ottmann, C; Combaret, V; Vigny, M; Kamaraj, S; Palmer, RH; Hallberg, B
Translational oncology
4
258-65
2011
Show Abstract
Activating mutations in the kinase domain of anaplastic lymphoma kinase (ALK) have recently been shown to be an important determinant in the genetics of the childhood tumor neuroblastoma. Here we discuss an in-depth analysis of one of the reported gain-of-function ALK mutations-ALK(I1250T)-identified in the germ line DNA of one patient. Our analyses were performed in cell culture-based systems and subsequently confirmed in a Drosophila model. The results presented here indicate that the germ line ALK(I1250T) mutation is most probably not a determinant for tumor initiation or progression and, in contrast, seems to generate a kinase-dead mutation in the ALK receptor tyrosine kinase (RTK). Consistent with this, stimulation with agonist ALK antibodies fails to lead to stimulation of ALK(I1250T) and we were unable to detect tyrosine phosphorylation under any circumstances. In agreement, ALK(I1250T) is unable to activate downstream signaling pathways or to mediate neurite outgrowth, in contrast to the activated wild-type ALK receptor or the activating ALK(F1174S) mutant. Identical results were obtained when the ALK(I1250T) mutant was expressed in a Drosophila model, confirming the lack of activity of this mutant ALK RTK. We suggest that the ALK(I1250T) mutation leads to a kinase-dead ALK RTK, in stark contrast to assumed gain-of-function status, with significant implications for patients reported to carry this particular ALK mutation. | | 21804922
|
Identification of oncogenic point mutations and hyperphosphorylation of anaplastic lymphoma kinase in lung cancer.
Wang, YW; Tu, PH; Lin, KT; Lin, SC; Ko, JY; Jou, YS
Neoplasia (New York, N.Y.)
13
704-15
2011
Show Abstract
The oncogenic property of anaplastic lymphoma kinase (ALK) plays an essential role in the pathogenesis of various cancers and serves as an important therapeutic target. In this study, we identified frequent intragenic loss of heterozygosity and six novel driver mutations within ALK in lung adenocarcinomas. Overexpression of H694R or E1384K mutant ALK leads to hyperphosphorylation of ALK, and activation of its downstream mediators STAT3, AKT, and ERK resulted in enhanced cell proliferation, colony formation, cell migration, and tumor growth in xenograft models. Furthermore, the activated phospho-Y1604 ALK was increasingly detected in 13 human lung cancer cell lines and 263 lung cancer specimens regardless of tumor stages and types. Treatment of two different ALK inhibitors, WHI-P154 and NVP-TAE684, resulted in the down-regulation of aberrant ALK signaling, shrinkage of tumor, and suppression of metastasis and significantly improved survival of ALK mutant-bearing mice. Together, we identified that novel ALK point mutations possessed tumorigenic effects mainly through hyperphosphorylation of Y1604 and activation of downstream oncogenic signaling. The upregulated phospho-Y1604 ALK could serve as a diagnostic biomarker for lung cancer. Furthermore, targeting oncogenic mutant ALKs with inhibitors could be a promising strategy to improve the therapeutic efficacy of fatal lung cancers. | Western Blotting | 21847362
|
Crystal-induced neutrophil activation. IX. Syk-dependent activation of class Ia phosphatidylinositol 3-kinase.
Popa-Nita, O; Rollet-Labelle, E; Thibault, N; Gilbert, C; Bourgoin, SG; Naccache, PH
Journal of leukocyte biology
82
763-73
2007
Show Abstract
The deposition of monosodium urate (MSU) crystals in the joints of humans leads to an extremely acute, inflammatory reaction, commonly known as gout, characterized by a massive infiltration of neutrophils. Direct interactions of MSU crystals with human neutrophils and inflammatory mediators are crucial to the induction and perpetuation of gout attacks. The intracellular signaling events initiated by the physical interaction between MSU crystals and neutrophils depend on the activation of specific tyrosine kinases (Src and Syk, in particular). In addition, PI-3Ks may be involved. The present study investigates the involvement of the PI-3K family in the mediation of the responses of human neutrophils to MSU crystals. The results obtained indicate that the interaction of MSU crystals with human neutrophils leads to the stimulation of class Ia PI-3Ks by a mechanism that is dependent on the tyrosine kinase Syk. We also found an increase in the amount of p85 associated with the Nonidet P-40-insoluble fraction derived from MSU crystal-stimulated human neutrophils. Furthermore, MSU crystals induce the formation of a complex containing p85 and Syk, which is mediated by the Src family kinases. Finally, evidence is also obtained indicating that the activation of PI-3Ks by MSU crystals is a critical element regulating phospholipase D activation and degranulation of human neutrophils. The latter response is likely to be involved in the joint and tissue damage that occurs in gouty patients. | | 17535983
|
The Mannich base NC1153 promotes long-term allograft survival and spares the recipient from multiple toxicities.
Stanislaw M Stepkowski, Judy Kao, Mou-Er Wang, Neelam Tejpal, Hemangshu Podder, Lucrezia Furian, Jonathan Dimmock, Amitabh Jha, Umashankar Das, Barry D Kahan, Robert A Kirken
Journal of immunology (Baltimore, Md. : 1950)
175
4236-46
2005
Show Abstract
JAK3 is a cytoplasmic tyrosine kinase with limited tissue expression but is readily found in activated T cells. Patients lacking JAK3 are immune compromised, suggesting that JAK3 represents a therapeutic target for immunosuppression. Herein, we show that a Mannich base, NC1153, blocked IL-2-induced activation of JAK3 and its downstream substrates STAT5a/b more effectively than activation of the closely related prolactin-induced JAK2 or TNF-alpha-driven NF-kappaB. In addition, NC1153 failed to inhibit several other enzymes, including growth factor receptor tyrosine kinases, Src family members, and serine/threonine protein kinases. Although NC1153 inhibited proliferation of normal human T cells challenged with IL-2, IL-4, or IL-7, it did not block T cells void of JAK3. In vivo, a 14-day oral therapy with NC1153 significantly extended survival of MHC/non-MHC mismatched rat kidney allografts, whereas a 90-day therapy induced transplantation tolerance (>200 days). Although NC1153 acted synergistically with cyclosporin A (CsA) to prolong allograft survival, it was not nephrotoxic, myelotoxic, or lipotoxic and did not increase CsA-induced nephrotoxicity. In contrast to CsA, NC1153 was not metabolized by cytochrome P450 3A4. Thus, NC1153 prolongs allograft survival without several toxic effects associated with current immunosuppressive drugs. | | 16177063
|
Regulation of ubiquitin protein ligase activity in c-Cbl by phosphorylation-induced conformational change and constitutive activation by tyrosine to glutamate point mutations.
Kassenbrock, CK; Anderson, SM
The Journal of biological chemistry
279
28017-27
2004
Show Abstract
c-Cbl down-regulates receptor tyrosine kinases by conjugating ubiquitin to them, leading to receptor internalization and degradation. The ubiquitin protein ligase activity of c-Cbl (abbreviated as E3 activity) is mediated by its RING finger domain. We show here that the E3 activity of c-Cbl is negatively regulated by other domains present in the amino-terminal half of the protein (the TKB and linker helix domains) and that this negative regulation is removed when the protein is phosphorylated on tyrosine residues. Protease digestion studies indicate that tyrosine phosphorylation alters the conformation of c-Cbl. We also show that mutation of certain conserved tyrosine residues to glutamate can constitutively activate the E3 activity of c-Cbl. In particular, a Y371E mutant shows constitutive E3 activity while retaining the ability to bind epidermal growth factor receptor (EGFR). The Y371E mutant also has altered protease sensitivity from wild type, instead resembling the proteolytic pattern seen with tyrosine-phosphorylated c-Cbl. Mutation of the homologous tyrosine residue in Cbl-b to glutamate also leads to E3 activation while retaining EGFR-binding ability. These studies argue that Tyr-371 plays a key role in activating the E3 activity of c-Cbl and that the Y371E mutant may partially mimic phosphorylation at that site. However, Tyr-371 point mutants of c-Cbl are still able to undergo phosphorylation-induced E3 activation, and we show that Tyr-368 can also be phosphorylated in addition to Tyr-371, and contributes to activation. | | 15117950
|
Insulin receptor substrate 4 associates with the protein IRAS.
Sano, H; Liu, SC; Lane, WS; Piletz, JE; Lienhard, GE
The Journal of biological chemistry
277
19439-47
2002
Show Abstract
The insulin receptor substrates (IRSs) are key components in signaling from the insulin receptor, and consequently any proteins that interact with them are expected to participate in insulin signaling. In this study we have searched for proteins that interact with IRS-4 by identifying the proteins that coimmunoprecipitated with IRS-4 from human embryonic kidney 293 cells by microsequencing through mass spectrometry. A group of proteins was found. These included phosphatidylinositol 3-kinase, a protein previously identified as an IRS-4 interactor, and several proteins for which there was no previous evidence of IRS-4 association. One of these proteins, named IRAS, that had been found earlier in another context was examined in detail. The results from the overexpression of IRAS, where its amount was about the same as that of IRS-4, indicated that IRAS associated directly with IRS-4 and showed that the increased complexation of IRS-4 with IRAS did not alter the insulin-stimulated tyrosine phosphorylation of IRS-4 or the association of IRS-4 with phosphatidylinositol 3-kinase or Grb2. On the other hand, overexpression of IRAS enhanced IRS-4-dependent insulin stimulation of the extracellularly regulated kinase. The domains of IRAS and IRS-4 responsible for the association of these two proteins were identified, and it was shown that IRAS also associates with IRS-1, IRS-2, and IRS-3. | | 11912194
|
Inhibition of Src family kinases blocks epidermal growth factor (EGF)-induced activation of Akt, phosphorylation of c-Cbl, and ubiquitination of the EGF receptor.
Kassenbrock, CK; Hunter, S; Garl, P; Johnson, GL; Anderson, SM
The Journal of biological chemistry
277
24967-75
2002
Show Abstract
Stimulation of T47D cells with epidermal growth factor (EGF) results in the activation of the intrinsic tyrosine kinases of the receptor and the phosphorylation of multiple cellular proteins including the receptor, scaffold molecules such as c-Cbl, adapter molecules such as Shc, and the serine/threonine protein kinase Akt. We demonstrate that EGF stimulation of T47D cells results in the activation of the Src protein-tyrosine kinase and that the Src kinase inhibitor PP1 blocks the EGF-induced phosphorylation of c-Cbl but not the activation/phosphorylation of the EGF receptor itself. PP1 also blocks EGF-induced ubiquitination of the EGF receptor, which is presumably mediated by phosphorylated c-Cbl. Src is associated with c-Cbl, and we have previously demonstrated that the Src-like kinase Fyn can phosphorylate c-Cbl at a preferred binding site for the p85 subunit of phosphatidylinositol 3'-kinase. PP1 treatment blocks EGF-induced activation of the anti-apoptotic protein kinase Akt suggesting that Src may regulate activation of Akt, perhaps by a Src --greater than c-Cbl --greater than phosphatidylinositol 3'-kinase --greater than Akt pathway. | Western Blotting | 11994282
|
Regulated secretion of neurotrophins by metabotropic glutamate group I (mGluRI) and Trk receptor activation is mediated via phospholipase C signalling pathways
Canossa, M., et al
Embo J, 20:1640-50 (2001)
2001
| Immunoprecipitation | 11285228
|